
Amgen 2018: Company-wide transformation
From 2014 through year-end 2017, Amgen delivered $1.5 billion in transformation and process improvement savings that the company has reinvested in launching new medicines, advancing the product pipeline, and pursuing external business development opportunities.
![]()
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, CA 91320-1799
Telephone: 805-447-1000
Website: amgen.com
Best-Selling Rx Products
| Product | 2017 Sales | 2017 Sales |
|---|---|---|
| Enbrel |
$5,433 |
$5,965 |
| Neulasta |
$4,534 |
$4,648 |
| Aranesp |
$2,053 |
$2,093 |
| Prolia | $1,968 | $1,635 |
| Sensipar/Mimpara | $1,718 | $1,582 |
| XGEVA | $1,575 | $1,529 |
| Epogen | $1,096 | $1,282 |
| Kyprolis | $835 | $692 |
| Vectibix | $642 | $611 |
| Nplate |
$642 |
$584 |
| Neupogen |
$549 |
$765 |
All sales are in millions of dollars.
Financial Performance
| 2017 | 2016 | |
|---|---|---|
| Revenue |
$22,849 |
$22,991 |
| Net income |
$1,979 |
$7,722 |
| Diluted EPS |
$2.69 |
$10.24 |
| R&D expense |
$3,562 |
$3,840 |
All figures are in millions of dollars, except EPS.
| 1H 2018 | 1H 2017 | |
|---|---|---|
| Revenue |
$11,613 |
$11,274 |
| Net income |
$4,607 |
$4,222 |
| Diluted EPS |
$6.73 |
$5.71 |
| R&D expense |
$1,629 |
$1,642 |
All figures are in millions of dollars, except EPS.
Amgen emerged from 2017 after generating company-record performances in revenue ($22.85 billion), non-GAAP earnings per share ($12.58), and free cash flow ($10.5 billion).
“Consistent with our belief in the long-term value of innovation, we invested $3.5 billion in research and development in support of a very promising pipeline of new medicines,” states Chairman, CEO and President Robert A. Bradway about the 2017 results. “Amgen delivered a one-year total shareholder return of 22 percent and a five-year return of 125 percent, outperforming the one- and five-year average total shareholder return of our peer group of 10% and 101%, respectively.”

Chairman, CEO & President Robert A. Bradway: “Amgen’s strong performance in the second quarter (2018) was driven by double-digit, volume-driven growth from our new and recently launched products. Our two most recently launched products, Aimovig and Parsabiv, are off to a strong start.”
More record-breaking figures are anticipated for Amgen for full-year 2018, including revenue and non-GAAP earnings per share.
Six therapeutic fields constitute the core of Amgen’s business: cardiovascular, oncology/hematology, neuroscience, inflammation, nephrology, and bone health. Executive leadership says in order to accomplish the aspiration of becoming the world’s best human therapeutics company, Amgen needs to aim high and take on tough challenges.
“As an example, we have seen a number of our peers abandon their efforts to develop new treatments for neurological disorders such as Alzheimer’s, a disease that afflicts 1 in 10 people age 65 and older,” according to Bradway. “Without a doubt, Alzheimer’s has proven to be an incredibly difficult target. Nonetheless, we remain in the fight. We are working on a variety of mechanisms in Alzheimer’s disease, including a molecule being tested in patients with a very high genetic risk for Alzheimer’s who have yet to develop cognitive symptoms of the disease – a promising new approach.”
During 2017, the company secured 80 marketing approvals for new products and new indications in more than 30 countries, which Amgen management says is a continuation of a significant shift in the product portfolio on two important dimensions.
“First, the balance of our portfolio is shifting to newer products with very favorable long-term growth potential,” Bradway says. “For example, three of our currently marketed products – Repatha, Prolia, and Kyprolis – can benefit significantly more patients than they do today. Together, these three products generated more than $3 billion in sales during 2017 and collectively grew by over 25% year-over-year.”
Those three products are on track in 2018 to exceed last year’s performance, with combined first-half sales coming in at $1.86 billion, representing a 27.1 percent increase versus their joint first-half 2017 total.
“Our portfolio will shift over time in another important respect – from medicines that predominantly serve small, ‘specialty’ patient populations to medicines that serve large patient populations,” Bradway notes. “This gives us the opportunity to make an even greater impact on health. Repatha and Prolia exemplify this shift, as do other medicines in our pipeline.”
Those other medicines include Aimovig, Amgen’s novel preventive treatment for migraine that won U.S. regulatory approval during May 2018. Aimovig represents the first FDA-approved preventive migraine treatment in a new class of drugs that work by blocking the activity of calcitonin gene-related peptide, a molecule that is involved in migraine attacks.
During the past five years, Amgen has expanded the company’s global presence from about 50 countries to 100 while attaining a stronger return on “considerable investments” in R&D and manufacturing. “One market of particular interest to us is China, home to one-fifth of the world’s population,” Bradway explains. “Our current and emerging portfolio of medicine matches up well with China’s top health challenges, one of which is cardiovascular disease. In 2018, we expect to launch our first medicine in China, Repatha. An estimated 450 million people in China have high cholesterol, so Repatha has the potential to have a profound impact on public health there.”
2018 PERFORMANCE & OUTLOOK
Revenue increased in first-half 2018 for Amgen by 3 percent compared to same-time 2017 to a total of $11.61 billion, driven primarily by higher unit demand. Net income improved 9 percent year-over-year to $4.61 billion during the 2018 first half, with diluted EPS rising 18 percent to $6.73 versus the amount reported for January-June 2017.
In announcing the company’s second-quarter results, management updated Amgen’s full-year 2018 guidance to total revenue in the range of $22.5 billion to $23.2 billion; previously, the revenue was forecast to land between $21.9 billion and $22.8 billion. On a GAAP basis, 2018 EPS is projected in the range of $11.83 to $12.62 (previously $11.30 to $12.28). On a non-GAAP basis, the 2018 EPS range is $13.30 to $14.00 (previously $12.80 to $13.70).
Amgen’s best-selling products during the first six months of 2018 were Enbrel ($2.41 billion, down 9 percent compared to first-half 2017), Neulasta ($2.26 billion, down 2 percent), Prolia ($1.1 billion, up 19 percent), Aranesp ($926 million, down 11 percent), Sensipar/Mimpara ($917 million, up 8 percent), XGEVA ($897 million, up 13 percent), and Epogen ($494 million, down 12 percent).

As one of Amgen’s top-selling medicines, once-daily Sensipar manages secondary hyperparathyroidism by lowering three key lab values in adult patients with secondary HPT on dialysis.
According to EvaluatePharma’s World Preview 2018 Report, Amgen is forecast during 2024 to rank 11th among all companies in terms of worldwide prescription drug sales at $24.8 billion and No. 4 in global Rx product sales from biotechnology at $22 billion.
According to the EP report, in terms of value creation from pipeline products launched from 2015 onward, Amgen ranks No. 5 among companies with total cumulative sales reaching $13.7 billion for the 2018-2024 forecast period, led by the migraine medicine Aimovig and the inflammatory disease treatment Amjevita. That forecast lists 20 lead products among the projected top 10 companies, with Amjevita representing the only biosimilar. “Amgen’s biosimilar portfolio in total is highly impactful on pipeline value creation with consensus forecasts for biosimilars representing 60 percent of pipeline value. This suggests that Amgen’s strategy of repurposing their biotechnology expertise to develop biosimilars is expected to be a success but also that the market expects limited revenue resulting from innovation,” according to EvaluatePharma analysis.
In terms of forecast global sales for 2024, the EvaluatePharma report projects that Amgen’s best-selling products will be Prolia and XGEVA with combined sales of nearly $6 billion, Enbrel with sales of $4.07 billion (including those contributed by marketers Pfizer and Takeda), and Repatha at $4.06 billion (including sales recorded by Astellas Pharma).
THERAPEUTIC PRODUCT APPROVALS/LAUNCHES & PIPELINE UPDATES DURING 2018
Aimovig won FDA marketing clearance in May for the preventive treatment of migraine in adults. The medicine is the only FDA-approved treatment specifically developed to prevent migraine by blocking the calcitonin gene-related peptide receptor (CGRP-R), which is believed to play a critical role in migraine. Aimovig 70 mg is self-administered once every month through Amgen’s device, the SureClick Autoinjector, and does not necessitate a loading dose.
Aimovig (erenumab-aooe) has been studied in several large worldwide, randomized, double-blind, placebo-controlled trials to evaluate the product’s efficacy and safety in migraine prevention. More than 3,000 patients have participated in the Aimovig clinical program across four placebo-controlled Phase II and Phase III clinical trials and their open-label extensions.
Amgen and Novartis revealed near the end of June new data from two open-label extension (OLE) studies showing long-term efficacy, safety, and tolerability of Aimovig in patients with chronic and episodic migraine. Data from a one-year study in chronic migraine patients reinforced the established safety and efficacy profile of Aimovig in long-term use. A three-year interim analysis from an ongoing five-year study of episodic migraine patients – the longest running study of a calcitonin gene-related peptide therapy – reinforced the long-term safety and tolerability of the drug. Aimovig is the first FDA-approved therapy targeting the CGRP pathway.
Amgen during April presented first-of-its-kind data at the American Academy of Neurology Annual Meeting reinforcing robust and consistent efficacy of Aimovig for migraine patients with multiple treatment failures. The LIBERTY study was carried out in patients who have tried two to four therapies without success – a uniquely difficult-to-treat population often excluded from migraine prevention trials. Patients taking the medication had nearly three-fold higher odds of having their migraine days reduced by half or more compared to placebo. Amgen says safety and tolerability were consistent with results seen in the pivotal clinical program; more than 97 percent of those taking Aimovig completed the double-blind treatment phase.
The FDA during June gave the green light for a supplemental new drug application to add the positive overall survival data from the Phase III ASPIRE study to the U.S. prescribing information for Kyprolis (carfilzomib). Data added to the label demonstrated that Kyprolis, lenalidomide and dexamethasone (KRd) significantly reduced the risk of death by 21 percent and extended overall survival by 7.9 months compared to lenalidomide and dexamethasone alone (Rd) in patients with relapsed or refractory multiple myeloma. Amgen reported on April 1 that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion for the same label variation.
Since Kyprolis’ first marketing approval during 2012, more than 80,000 patients around the globe have received the multiple myeloma drug.
Initially cleared for U.S. marketing during 2012, more than 80,000 patients around the globe have received Kyprolis. The product is FDA-approved in combination with dexamethasone or with lenalidomide plus dexamethasone for treating patients with relapsed or refractory multiple myeloma who have received one to three lines of therapy; and as a single agent for treating patients with relapsed or refractory multiple myeloma who have received one or more lines of therapy.
Kyprolis is available in the European Union for use in combination with dexamethasone or with lenalidomide plus dexamethasone for treating patients with relapsed or refractory multiple myeloma who have received at least one prior therapy.
Kyprolis is additionally approved in Argentina, Australia, Bahrain, Canada, Hong Kong, Israel, Japan, Kuwait, Lebanon, Macao, Mexico, Thailand, Colombia, South Korea, Canada, Qatar, Switzerland, United Arab Emirates, Turkey, Russia, Brazil, India, and Oman. Other regulatory applications for the medicine have been filed with health authorities.
Proteasomes play a significant role in cell function and growth by breaking down proteins that are damaged or no longer needed. Kyprolis has been demonstrated to block proteasomes, resulting in an excessive build-up of proteins within cells. In certain cells, the drug can cause cell death, particularly in myeloma cells because they are more likely to contain a higher amount of abnormal proteins.
During August, Amgen announced the filing of a supplemental new drug application with U.S. regulators to expand the prescribing information for Kyprolis to include a once-weekly dosing option in combination with dexamethasone (Kd) for patients with relapsed or refractory multiple myeloma. The sNDA is based on data from the Phase III A.R.R.O.W. study showing that Kyprolis administered once weekly at 70 mg/m2 with dexamethasone (once-weekly Kd) achieved superior progression-free survival and overall response rates, with a comparable safety profile compared to twice-weekly Kyprolis at 27 mg/m2 and dexamethasone (twice-weekly Kd) in patients with relapsed and refractory multiple myeloma.
During January, the FDA approved an sNDA for the addition of overall survival (OS) data to the Kyprolis label from the Phase III head-to-head ENDEAVOR study. Also in January, the CHMP issued a positive opinion recommending a label variation for Kyprolis to include the updated OS data from ENDEAVOR in patients with relapsed or refractory multiple myeloma (Kyprolis and dexamethasone versus Velcade [bortezomib] and dexamethasone [Vd]). The ENDEAVOR study showed that Kd reduced the risk of death by 21 percent and increased OS by 7.6 months compared to Vd in patients with relapsed or refractory multiple myeloma.
U.S. regulators approved an expanded indication of Prolia (denosumab) on May 18: for treating osteoporosis associated with newly initiating or sustained systemic glucocorticoid therapy in men and women at high risk of fracture. The drug was previously cleared by the FDA for four other indications: treating postmenopausal women with osteoporosis at high risk for fracture; as a treatment to increase bone mass in men with osteoporosis at high risk for fracture; as a treatment to increase bone mass in men at high risk for fracture receiving androgen deprivation therapy for nonmetastatic prostate cancer; and as a treatment to increase bone mass in women at high risk for fracture receiving adjuvant aromatase inhibitor therapy for breast cancer.
During second-quarter 2018, the U.S. FDA and European Commission approved a new indication for the blockbuster brand Prolia: for the treatment of glucocorticoid-induced osteoporosis, the most common drug-induced form of the disease.
Prolia during June received the drug’s third approved indication in Europe: for the treatment of bone loss associated with long-term systemic glucocorticoid therapy in adult patients at increased risk of fracture. The European Commission approval is based on the positive results of a Phase III trial that assessed the safety and efficacy of Prolia versus risedronate in patients receiving glucocorticoid treatment. The drug was previously granted EU approval for treating osteoporosis in postmenopausal women and in men at increased risk of fractures, and for treating bone loss associated with hormone ablation in men with prostate cancer at increased risk of fractures.
Prolia is the first approved therapy that specifically targets RANK ligand, an essential regulator of bone-removing cells known as osteoclasts. The medication is approved and marketed in more than 80 countries.
Blincyto during March became the first- FDA-approved therapy for minimal residual disease (MRD), which refers to the presence of cancer cells that remain detectable despite a patient’s having achieved complete remission by conventional assessment. MRD is only measurable via the use of highly sensitive testing methods that detect cancer cells in the bone marrow with a sensitivity of at least one cancer cell in 10,000 cells – compared to about one in 20 with a conventional microscope-based evaluation. The supplemental Biologics License Application was cleared for the treatment of adults and children with B-cell precursor acute lymphoblastic leukemia (ALL) in first or second complete remission with MRD greater than or equal to 0.1 percent. The accelerated approval was based on data from the Phase II BLAST trial, the largest prospective study in minimal residual disease-positive ALL.
The European Commission near the end of August cleared for marketing an expanded indication for Blincyto (blinatumomab) as monotherapy for treating pediatric patients aged 1 year or older with Philadelphia chromosome-negative CD19 positive B-cell precursor ALL, which is refractory or in relapse after receiving at least two prior therapies or in relapse after receiving prior allogeneic hematopoietic stem cell transplantation. The approval via the centralized procedure is valid in all EU and European Economic Area-European Free Trade Association states (Norway, Iceland and Liechtenstein). The marketing clearance is based on results from the Phase I/II ‘205 trial, an open-label, multicenter, single-arm study that evaluated the efficacy and safety of Blincyto in pediatric patients with relapsed or refractory B-cell precursor ALL.
Blincyto was initially approved in the EU during November 2015 for treating adults with Ph- relapsed or refractory B-cell precursor ALL. In June 2018, the European Commission granted a full marketing authorization for Blincyto based on the overall survival data from the Phase III TOWER trial in adult patients with Philadelphia chromosome-negative (Ph-) relapsed or refractory B-cell precursor ALL.
Blincyto represents the first bispecific T cell engager immunotherapy construct approved worldwide. Blincyto is additionally the first immunotherapy from Amgen’s BiTE platform, an innovative approach that helps the body’s immune system target cancer cells.
BiTE antibody constructs are a novel immune-oncology technology that can be engineered to target any tumor antigen expressed by any form of cancer. The modified antibodies are designed to kill malignant cells using a patient’s own immune system by bridging T cells to tumor cells. BiTE antibody constructs aid in connecting T cells to the targeted cell, with the intent of causing T cells to inject toxins which trigger cancer cell death (apoptosis). Amgen is developing BiTE antibody constructs to uniquely (or specifically) target numerous hematologic malignancies as well as solid tumors.
A supplemental Biologics License Application for Amgen’s XGEVA (denosumab) was approved by the FDA on January 5. The product’s U.S. indication was expanded to include the prevention of skeletal-related events in patients with bone metastases from solid tumors to include patients with multiple myeloma. XGEVA was already indicated in the U.S. for treating adults and skeletally mature adolescents with giant cell tumor of bone that is unresectable or where surgical resection is likely to result in severe morbidity, and for treating hypercalcemia of malignancy refractory to bisphosphonate therapy.
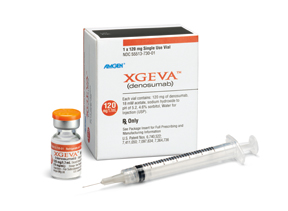
The fully human monoclonal antibody XGEVA targets the RANK ligand pathway to prevent the formation, function and survival of osteoclasts, which break down bone.
XGEVA was also the recipient of a European Commission approval, announced on April 2, for an expanded indication for the prevention of skeletal-related events in patients with multiple myeloma. The EU-approved indication now covers patients with bone metastases from solid tumors as well as those with multiple myeloma. The EU approval was based on the largest international study ever performed for the prevention of skeletal-related events in multiple myeloma patients. Additional regulatory filings for the product for the prevention of skeletal-related events in patients with multiple myeloma are under way and have been submitted to health authorities around the globe. XGEVA is additionally indicated in the EU for treating adults and skeletally mature adolescents with giant cell tumor of bone that is unresectable or where surgical resection is likely to result in severe morbidity.
XGEVA represents the first fully human monoclonal antibody that binds to and neutralizes RANK ligand – the protein essential for the formation, function and survival of osteoclasts – thereby inhibiting osteoclast-mediated bone destruction.
During February, Amgen announced results from the XGEVA Phase III ‘482 study – the largest international multiple myeloma trial for the prevention of skeletal-related events ever conducted (n=1,718) – were published in The Lancet Oncology. In this study, the product successfully met the primary endpoint, showing non-inferiority to zoledronic acid in delaying the time to first on-study skeletal-related event in patients with multiple myeloma (HR=0.98, 95 percent CI: 0.85-1.14).
Parsabiv (etelcalcetide), a treatment for secondary hyperparathyroidism in hemodialysis patients, was launched in the United States during first-quarter 2018 and generated U.S. sales of $102 million through the end of June.
Amgen gained European Commission clearance during May for Repatha (evolocumab) to prevent heart attack and stroke in adults with established cardiovascular disease. The European Commission approved the new indication in the Repatha label for adults with established atherosclerotic cardiovascular disease (myocardial infarction, stroke or peripheral arterial disease) to reduce cardiovascular risk by lowering low-density lipoprotein cholesterol (LDL-C) levels.
The cholesterol-lowering medicine Repatha is approved in more than 60 countries, including the U.S., Japan, Canada and all 28 member countries of the European Union.
During June, Amgen revealed new data demonstrating that Repatha significantly reduced LDL-C and non-high density lipoprotein cholesterol (non-HDL-C) in patients with Type 2 diabetes and hypercholesterolemia or mixed dyslipidemia, taking the maximum tolerated dose of moderate/high-intensity statin therapy. The dedicated study BANTING assessed the efficacy of Repatha (monthly dosing) in lowering LDL-C and improving other lipid levels in patients with Type 2 diabetes.
The human monoclonal antibody Repatha inhibits proprotein convertase subtilisin/kexin type 9 (PCSK9). The medicine binds to PCSK9 and inhibits circulating PCSK9 from binding to the low-density lipoprotein (LDL) receptor (LDLR), preventing PCSK9-mediated LDLR degradation and permitting LDLR to recycle back to the liver cell surface. By inhibiting the binding of PCSK9 to LDLR, Repatha increases the amount of LDLRs available to clear LDL from the blood, thereby reducing LDL-C levels.
Repatha has been granted marketing clearance in more than 60 countries, including the U.S., Japan, Canada and in all 28 countries that are members of the European Union. Regulatory submissions in other countries are pending.
Amgen and UCB announced during July the resubmission of the Biologics License Application to the FDA for Evenity (romosozumab). The investigational monoclonal antibody is intended for treating osteoporosis in postmenopausal women at high risk for fracture. The product increases bone formation and reduces bone resorption simultaneously to increase bone mineral density (BMD) and reduce the risk of fracture.
The re-filed BLA adds results from two more recent pivotal Phase III trials: ARCH, an alendronate-active comparator study including 4,093 postmenopausal women with osteoporosis who experienced a fracture, and BRIDGE, including 245 men with osteoporosis. U.S. regulators are evaluating the clinical benefit:risk profile of Evenity, including the cardiovascular safety signal seen in the ARCH trial, for the potential to reduce the risk of fractures and increase BMD in postmenopausal women with osteoporosis. The original FDA filing included data from a comprehensive Phase I and Phase II program and the Phase III placebo-controlled FRAME trial, including 7,180 postmenopausal women with osteoporosis.
An investigational bone-forming monoclonal antibody, Evenity has not yet been approved by any regulatory authority for treating osteoporosis. The European Medicines Agency and the Pharmaceuticals and Medical Device Agency in Japan are reviewing marketing applications. The MAA was accepted for review by the EMA in January.
Evenity is designed to work by inhibiting the activity of sclerostin, which enables the drug to rapidly increase bone formation and reduce bone resorption simultaneously. Evenity has been studied for the potential to reduce the risk of fractures in an extensive worldwide Phase III program, including two large fracture studies comparing Evenity to either placebo or active comparator in more than 11,000 postmenopausal women with osteoporosis. Amgen and UCB are jointly developing the new drug candidate.
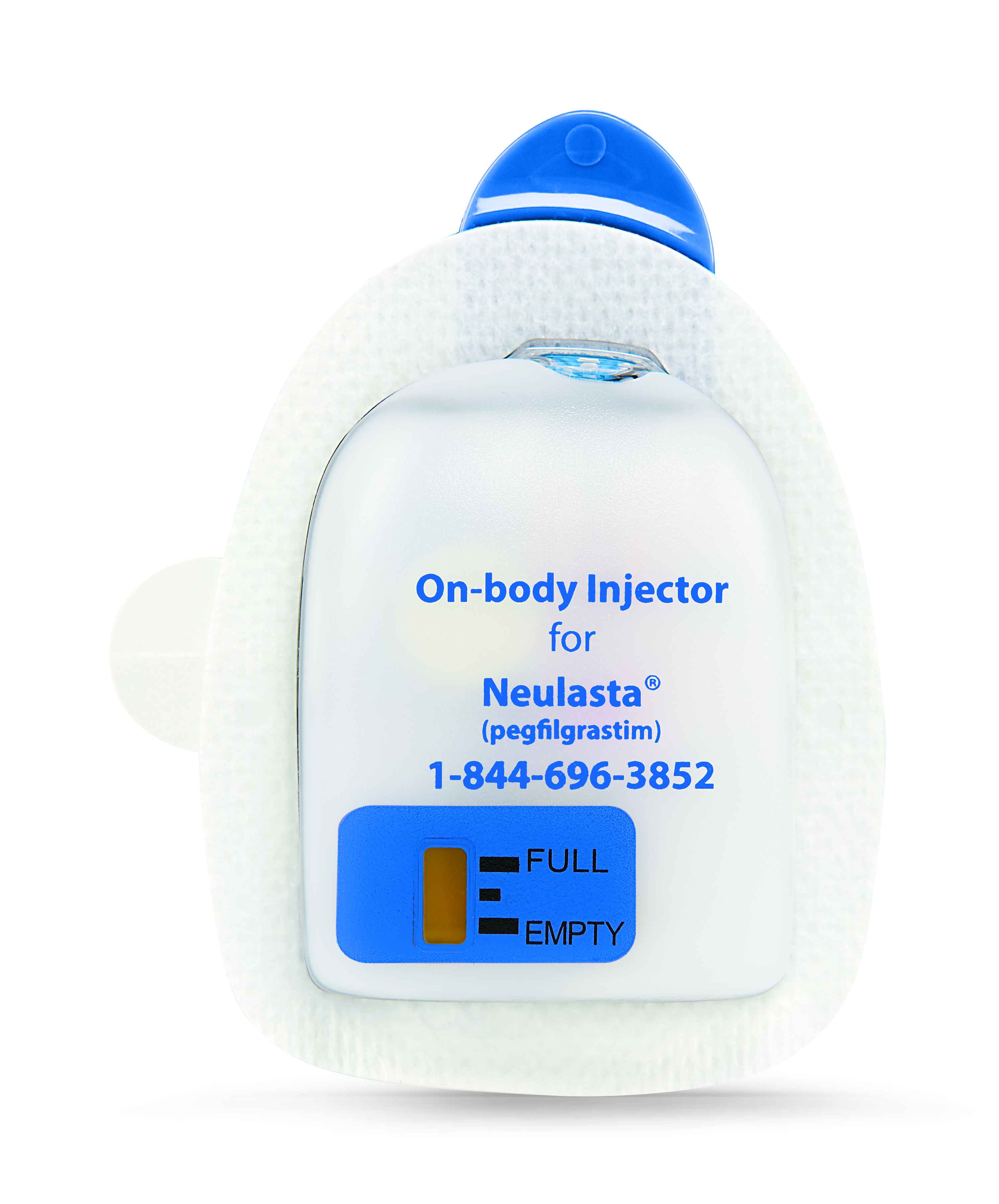
The Committee for Medicinal Products for Human Use of the European Medicines Agency during February 2018 issued a positive opinion recommending a label variation for Neulasta (pegfilgrastim) to include the Neulasta Onpro Kit.
Since 2004, Amgen and UCB have worked together under a collaboration and license deal to research, develop, and market antibody products targeting the protein sclerostin. As part of this agreement, both companies continue to collaborate on the development of romosozumab for treating osteoporosis. This gene-to-drug project shows how Amgen and UCB are joining forces to translate a genetic discovery into a new drug, turning conceptual science into a reality.
The EMA’s CHMP adopted a positive opinion in February recommending a label variation for Neulasta (pegfilgrastim) to include the Neulasta Onpro Kit. The Neulasta Onpro Kit brings together the efficacy of Neulasta with an innovative on-body injector delivery system. Neulasta was cleared for marketing in the European Union during 2002 for the reduction in the duration of neutropenia and the incidence of febrile neutropenia in adult patients treated with cytotoxic chemotherapy for malignancy (with the exception of chronic myeloid leukemia and myelodysplastic syndromes).
The former blockbuster Neulasta was also approved for use in the United States during 2002 and is indicated to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with non-myeloid malignancies receiving myelosuppressive anti-cancer drugs associated with a clinically significant incidence of febrile neutropenia. Neulasta is additionally indicated in the U.S. to increase survival in patients acutely exposed to myelosuppressive doses of radiation.
Amgen and AstraZeneca during September reported that the U.S. FDA granted Breakthrough Therapy Designation for tezepelumab in patients with severe asthma without an eosinophilic phenotype. The designation is supported by the tezepelumab Phase IIb PATHWAY data. The study demonstrated a significant reduction in the annual asthma exacerbation rate versus placebo in a broad population of severe asthma patients independent of baseline blood eosinophil count or other type 2 (T2) inflammatory biomarkers. Currently marketed biologic therapies only target T2 driven inflammation. The potential first-in-class medicine tezepelumab blocks thymic stromal lymphopoietin (TSLP) – an upstream modulator of multiple inflammatory pathways.
Tezepelumab is undergoing development in the Phase III PATHFINDER study program. The new product candidate is being developed by AstraZeneca in collaboration with Amgen.
BIOSIMILAR PROGRESS DURING 2018
Amgen Biosimilars is dedicated to building upon experience in the development and manufacturing of innovative human therapeutics to expand the company’s reach to patients with serious illnesses. According to management, biosimilars will help to maintain Amgen’s commitment to connect patients with vital medicines, and Amgen is well positioned to leverage the company’s four decades of biotech experience to create high-quality biosimilars and reliably supply them to patients worldwide. The portfolio consists of 10 products, including two marketed in the United States and three approved in the European Union.
Amgen and Allergan are collaborating on the development and commercialization of several oncology biosimilars. In December 2011, Amgen and Allergan (then known as Watson Pharmaceuticals) forged a collaboration to develop and commercialize on a global basis four oncology antibody biosimilar medicines. Amgen will assume primary responsibility for developing, manufacturing, and initially commercializing the oncology biosimilars.
The two companies received marketing authorization from the European Commission for MVASI (biosimilar bevacizumab) during January. MVASI represents the first biosimilar bevacizumab approved by the European Commission and is marketed for treating certain types of cancers. MVASI is the first targeted cancer biosimilar from Amgen’s portfolio to be granted approval in Europe. This is the first biosimilar from the Amgen-Allergan collaboration to win EU marketing authorization.
In September 2017, MVASI became the first anti-cancer biosimilar and the first biosimilar bevacizumab to receive U.S. marketing clearance. Bevacizumab is a recombinant immunoglobulin G1 (IgG1) monoclonal antibody (mAb) that binds to vascular endothelial growth factor (VEGF) and inhibits the interaction of VEGF with its receptors, VEGF receptor-1 and VEGF receptor-2, thus inhibiting establishment of new blood vessels needed for the maintenance and growth of solid tumors. Bevacizumab is marketed by Roche/Genentech as the blockbuster brand Avastin.
The European Commission in May granted marketing authorization for Kanjinti, a biosimilar to the blockbuster Herceptin (trastuzumab), for treating HER2-positive metastatic breast cancer, HER2-positive early breast cancer and HER2-positive metastatic adenocarcinoma of the stomach or gastroesophageal junction.
Also known by the product code ABP 980, Kanjinti represents a biosimilar version of Herceptin, which is a recombinant DNA-derived humanized monoclonal immunoglobulin G1 kappa antibody marketed worldwide for treating HER2-overexpressing early breast cancer, adjuvant breast cancer, metastatic breast cancer and metastatic gastric cancer.
Amgen and Allergan filed a Biologics License Application to the U.S. FDA for ABP 980 during 2017. During May 2018, Amgen received a complete response letter from the U.S. regulatory agency on the BLA.
Amgen during June reported results from a Phase III trial assessing the efficacy and safety of the biosimilar candidate ABP 710 versus the blockbuster brand Remicade (infliximab) in patients with moderate to severe rheumatoid arthritis. The results confirm non-inferiority versus infliximab but could not rule out superiority based on ABP 710’s primary efficacy endpoint, which compared the response difference measured by 20 percent or greater improvement defined by the American College of Rheumatology (ACR) Criteria, ACR20, at week 22.
ABP 710 is being developed as a biosimilar to the anti-tumor necrosis factor alpha monoclonal antibody infliximab, which is available in the United States, EU and other regions for treating moderate to severe rheumatoid arthritis, chronic severe plaque psoriasis, moderate to severe Crohn’s disease, moderate to severe ulcerative colitis, psoriatic arthritis and ankylosing spondylitis.

