
R&D/Pipelines Report 2022: Innovating to turn the tide
Innovating to turn the tide
By Andrew Humphreys • [email protected]
The world’s leading COVID-19 vaccine and therapeutic manufacturers continue to develop treatments for the waves of variants as well as life-changing therapies for disease areas outside the world of coronaviruses. The world’s efforts to combat the global pandemic continue to evolve, as does Coronavirus Disease 2019 as variants and subvariants constantly toss volleys of grenades at the battlefront lines. As of February 4, 2022, there were nearly 1,440 drugs and vaccines in development targeting COVID-19, according to Statista data. As of February 6, the amount of COVID-19 vaccine doses administered in the United States broke down as follows per Statista: 319,082,480 for Pfizer–BioNTech; 205,409,451 for Moderna; 18,246,402 for Johnson & Johnson/Janssen; and 481,711 for other vaccine manufacturers.
Pfizer and BioNTech have forecast that their COVID-19 vaccine will generate sales of $29 billion in 2022, after having produced an estimated $36 billion during 2021. As of January 10, Moderna had inked 2022 advance purchase agreements (APAs) for product sales of $18.5 billion (up from $17 billion announced during November 2021) and $3.5 billion in options including for any potential updated COVID-19 vaccine booster candidates. Moderna continues to have active discussions for additional 2022 COVID-19 vaccine contracts. Moderna sales for 2021 totaled $17.5 billion (unaudited).
Whereas the Pfizer-BioNTech and Moderna vaccines were granted Emergency Use Authorization (EUA) during December 2020, the J&J/Janssen vaccine EUA came on February 27, 2021. J&J reported COVID-19 vaccine sales of $2.39 billion for 2021, falling short of the reported target of $2.5 billion. Johnson & Johnson on January 25 predicted that 2022 COVID-19 vaccine sales would range from $3 billion to $3.5 billion, topping Wall Street forecasts, which had been lowered due to safety concerns and low U.S. uptake. In December 2021, the U.S. Centers for Disease Control and Prevention recommended the Pfizer-BioNTech or Moderna shots over J&J’s vaccine because of the risk of blood clots linked to Johnson & Johnson’s jab.
Development activity during the past two years has been dominated by vaccines and therapies targeted at ending the pandemic. Clarivate analysts say while not all have succeeded to market, the learnings accumulated during this time have been folded into other programs, both for COVID-19 and therapeutic areas such as oncology and other infectious diseases, including the flu and HIV. “Pharma’s battle against COVID-19 continues, with hundreds of vaccines and treatments in development even as some markets look to transition from pandemic to endemic disease mitigation,” the analysts say.
PFIZER & BIONTECH
“Pfizer and BioNTech co-developed the world’s first mRNA vaccine, providing a well-tolerated and effective tool to help address COVID-19 – the most devastating pandemic in a century – and demonstrating consistent, agile and high-quality manufacturing on an unprecedented scale,” said Mikael Dolsten, M.D., Ph.D., Chief Scientific Officer and President, Worldwide Research, Development & Medical, Pfizer.
Comirnaty became the first COVID-19 vaccine to gain U.S. FDA full approval.
Known by the trade name Comirnaty, it was the first COVID-19 vaccine to win approval from the U.S. Food and Drug Administration for ages 16 years and up, eight months after the FDA EUA was granted to Pfizer and BioNTech. The authorization was expanded to include individuals ages 12 through 15 years on May 10, 2021. Pfizer and BioNTech received the first FDA EUA of a COVID-19 vaccine booster on September 22, 2021, granted for individuals 65 years of age and older as well as ages 18 through 64 within certain high-risk groups. The companies were issued the first U.S. EUA of a COVID-19 vaccine in children ages 5-11 years on October 29, 2021. An expanded EUA to include people 18 years and older was issued on November 19, 2021. FDA cleared the EUA for the vaccine booster for patients 16 years and older on December 9, 2021. The first Emergency Use Authorization in the United States for a COVID-19 vaccine booster in adolescents 12 through 15 years of age was granted to the companies on January 3, 2022.
Pfizer and BioNTech on February 1, 2022, initiated a rolling submission for Emergency Use Authorization of the companies’ COVID-19 Vaccine for children 6 months through 4 years of age following a request from the U.S. FDA. With pediatric COVID-19 cases surpassing 10 million and at the request of the U.S. regulatory agency, the companies submitted available data on the safety and efficacy of two 3 µg doses as part of a three-dose primary series for this age group to address the urgent public health need. Pfizer and BioNTech intend to submit additional data on a third 3 µg dose in this age group during 2022.If FDA authorization is granted, the Pfizer-BioNTech vaccine would be the first COVID-19 vaccine available for pediatric populations under the age of 5 years.
Pfizer and BioNTech announced on January 25, 2022, a clinical trial to assess the safety, tolerability and immunogenicity of an Omicron-based vaccine candidate in healthy adults 18 through 55 years of age. The study will have three cohorts evaluating different regimens of the current Pfizer-BioNTech COVID-19 vaccine Comirnaty or an Omicron-based vaccine. The study will draw upon some participants from the companies’ Phase III COVID-19 booster trial and is part of their continuing efforts to address Omicron and determine the potential necessity for variant-based vaccines. The first participants enrolled in the trial received the Omicron-based vaccine candidate as a two-dose primary series and as a booster dose.
Clinical and real-world data continue to find individuals who are vaccinated, particularly those who have received a booster, maintain a high level of protection against Omicron, especially against severe disease and hospitalization. Pfizer and BioNTech previously announced that they expect to produce 4 billion doses of their COVID-19 vaccine during 2022, and this capacity is not expected to change if an adapted vaccine is necessary.
Pfizer and BioNTech on January 24 announced the publication of new results from two laboratory studies showing that three doses of Comirnaty (BNT162b2) elicited antibodies that neutralize the Omicron variant, B.1.1.529. Data sets published in the peer-reviewed journal Science confirm previously announced initial study results showing that serum antibodies induced by BNT162b2 neutralize the SARS-CoV-2 Omicron variant after immunization with three doses. In comparison, sera from participants who received two doses of the vaccine revealed limited neutralization titers against the Omicron variant in each data set, indicating that two doses of BNT162b2 may not be sufficient to protect against infection with the new variant. However, based on observations that around 85 percent of epitopes in the spike protein recognized by CD8+ T cells are not affected by the mutations in the Omicron variant, Pfizer and BioNTech believe two doses may still induce protection against severe disease.
In a non-COVID-related deal between the two companies, in early January 2022 Pfizer and BioNTech struck a new research, development and commercialization collaboration to develop a potential first messenger RNA (mRNA)-based vaccine for the prevention of shingles (herpes zoster virus, or HZV). The debilitating, disfiguring and painful disease impacts about one in three people in the United States during their lifetime. This is the third collaboration between the two companies in the infectious diseases field, following the influenza vaccine collaboration initiated during 2018 and the COVID-19 vaccine collaboration started in 2020.
The shingles product candidates will be based on BioNTech’s proprietary mRNA technology and on Pfizer’s antigen technology. Clinical studies are anticipated by the companies to begin during the second half of 2022.
In addition to the company’s highly successful COVID-19 vaccine, Pfizer has developed an oral antiviral for the treatment of COVID-19. On December 22, the U.S. Food and Drug Administration issued an EUA for Paxlovid (nirmatrelvir tablets and ritonavir tablets, co-packaged for oral use) for treating mild-to-moderate coronavirus disease in adults and pediatric patients (12 years of age and older weighing at least 40 kilograms or about 88 pounds) with positive results of direct SARS-CoV-2 testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.
“Today’s authorization introduces the first treatment for COVID-19 that is in the form of a pill that is taken orally – a major step forward in the fight against this global pandemic,” noted Patrizia Cavazzoni, M.D., Director of the FDA’s Center for Drug Evaluation and Research. “This authorization provides a new tool to combat COVID-19 at a crucial time in the pandemic as new variants emerge and promises to make antiviral treatment more accessible to patients who are at high risk for progression to severe COVID-19.”
Pfizer was a global force in the biopharma world for many years before the company became a leader in the worldwide effort to combat the COVID-19 pandemic. Pfizer’s revolutionary medicines allow the company to enrich and extend life for people suffering from all types of diseases. Pfizer’s pipeline program included 94 assets as of November 2, 2021, including 38 in late-stage development or registration. Pfizer partners with thousands of study sites and tens of thousands of clinical trial participants around the globe that lead to life-changing medicines. Areas of focus include rare diseases, internal medicine, inflammation & immunology, vaccines, oncology, and anti-infectives.
Dolsten told Med Ad News, “I’ve never felt more confident about our pipeline. Even beyond the COVID-19 vaccine and our efforts to advance multiple investigational therapeutic options, we’ve continued great momentum across our pipeline. The extraordinary work done by our vaccine team was not a matter of chance, but a result of the 10-year journey we took at Pfizer to dramatically turn around our R&D organization. Over the last decade, we’ve been taking a hard look at our productivity, sharpening our focus, and implementing gradual changes across multiple dimensions.”
Dolsten continued, “From our deep understanding of biology, doubling down on exciting new modalities, and the efforts to empower our scientists for critical decision-making on their programs, our pipeline reflects the scientific opportunity partly brought about by the changes we’ve made.”
Pfizer: Advancing mRNA strategic development
v365
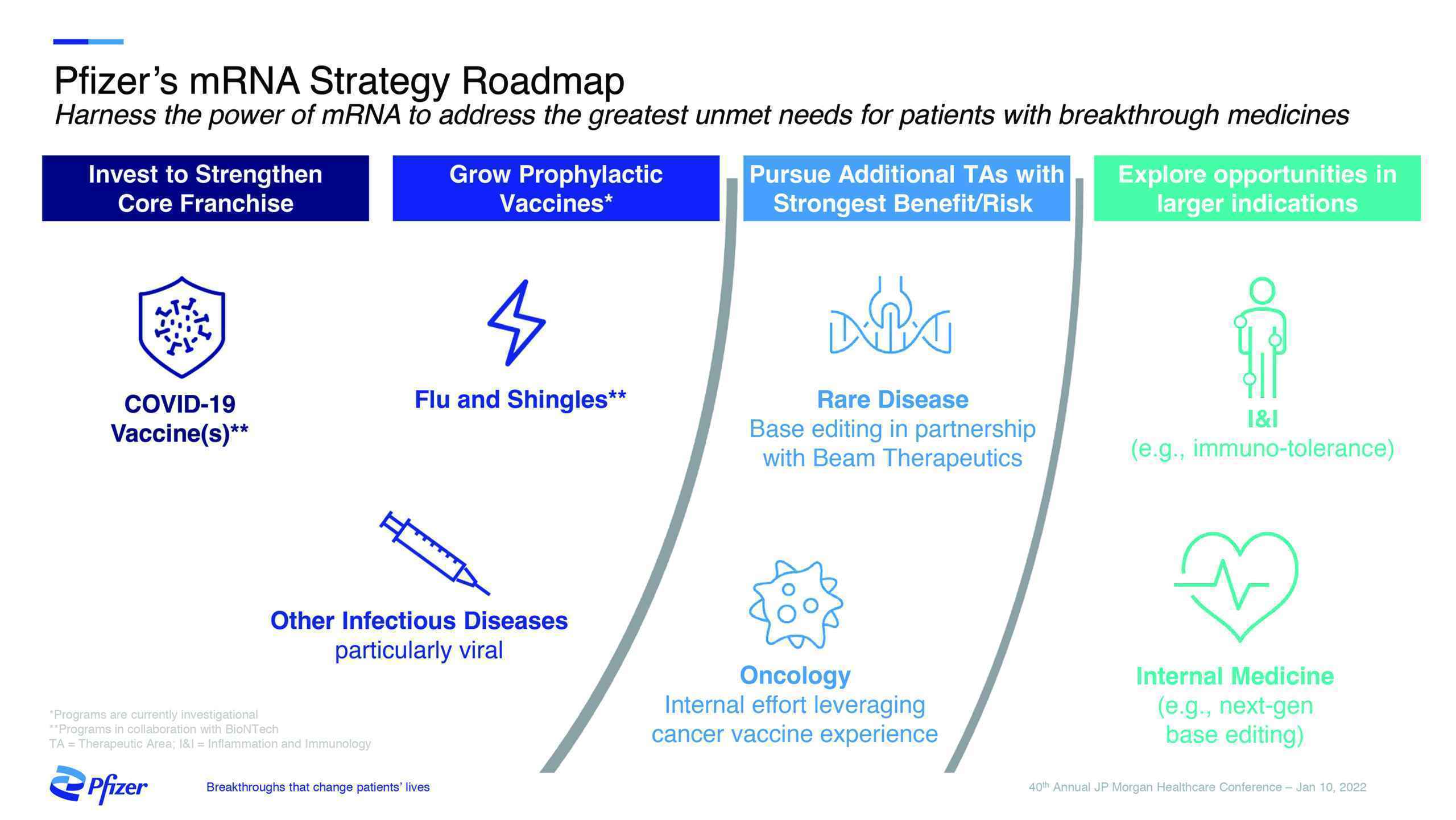
mRNA is expected to be one of Pfizer’s sustainable business drivers going forward, adding another potential treatment modality to the company’s armamentarium for vaccines and therapeutics, across its areas of focus. According to management, Pfizer’s Lightspeed methodology and internal cultural changes resulted in the acceleration of the platform’s development in recent years. Pfizer intends to continue to harness the power of mRNA to address the greatest patient challenges and contribute substantially to the company’s growth targets in the years ahead.
The first Pfizer internal mRNA experts were hired during September 2015. Pfizer and BioNTech inked a deal for an mRNA flu collaboration in August 2018. The two companies agreed on their COVID-19 collaboration as the pandemic took hold in the United States during March 2020. By December of that year, BNT162b2 received the vaccine’s first Emergency Use Authorizations in adults from the UK MHRA and U.S. FDA. Comirnaty would go on to become the first COVID-19 vaccine to gain FDA BLA approval in August 2021. The first patient was dosed in Pfizer’s mRNA flu vaccine in Phase I during September 2021. By the end of 2021, more than 3 billion doses of Comirnaty had been produced with 2.6 billion doses shipped, including 1 billion to low-income and middle-income countries.
To advance Pfizer’s mRNA strategy, the company has pursued four agreements aligned with its strategic priorities as announced during January 2022 with the following: BioNTech (collaboration agreement to jointly develop an mRNA Shingles (HZV) vaccine); a collaboration with Beam Therapeutics for three rare diseases (CNS, muscular, liver); Acuitas Therapeutics (a collaboration and option to non-exclusively license LNP technology for up to 10 targets); and Codex DNA (a research collaboration/license for synthetic DNA technology).
According to the New York-based company, these strategic collaborations provide many of the key enabling technologies necessary to deepen and expand Pfizer’s mRNA development. The BioNTech shingles collaboration adds a potential third mRNA viral vaccine candidate to Pfizer’s portfolio. Beam expands the use of mRNA into the area of rare diseases with the opportunity for precise gene editing. The Acuitas LNP collaboration/option/license provides potential LNPs for other future vaccine and therapeutics candidates. In terms of platform capabilities, Codex DNA’s synthetic DNA assembly could enable reduced production cycle times.
In the global collaboration agreement announced on January 5 to develop the first mRNA-based shingles vaccine product candidates, they will be based on BioNTech’s proprietary mRNA technology and on Pfizer’s antigen technology. Clinical studies are anticipated to begin during the second half of 2022. BioNTech received $225 million upfront and is eligible to receive future approval and sales milestone payments amounting up to $200 million as well as a share of gross profits arising from future product sales. Pfizer received an upfront payment of $25 million from BioNTech for the proprietary antigen sequences identified by Pfizer.
“With this agreement, we continue on our journey of discovery together, by advancing mRNA technology to tackle another health challenge ripe for scientific innovation, supported by our world-class manufacturing network,” Dolsten stated.
The four-year research collaboration agreed upon on January 10 between Pfizer and Beam combines Pfizer’s deep experience in worldwide drug development, including programs using mRNA, lipid nanoparticles (LNP) and gene therapy, with Beam’s leadership in base editing and mRNA/LNP delivery technologies. Beam received an upfront payment of $300 million, and is eligible to receive future milestone payments of up to $1.05 billion for a potential total consideration of up to $1.35 billion. Beam may opt into a global co-development and co-commercialization deal for one program. The collaboration’s R&D activities aim to advance potentially transformative therapies for patients living with rare genetic diseases.
In another transaction announced on January 10, a development and option agreement was reached under which Pfizer will have the option to license, on a non-exclusive basis, Acuitas’ LNP technology for up to 10 targets for vaccine or therapeutic development. A company concentrated on developing lipid nanoparticle delivery systems to enable messenger RNA-based therapeutics, Acuitas’ clinically validated LNP technology is used in the COVID vaccine Comirnaty.
“Our swift delivery of the world’s first mRNA-LNP-based vaccine made clear the promise of mRNA-LNP technology,” Dolsten said. “We are making significant investments to harness the power of the mRNA-LNP technology and deliver potential new breakthrough vaccines and therapeutics that address significant unmet needs for patients. This agreement expands our LNP capabilities and allows us to explore more projects within our existing vaccines area and new therapeutic areas where mRNA-LNP technology holds potential for success.”
Pfizer and Codex DNA agreed on January 10 to a strategic, multiyear, early access research collaboration leveraging Codex DNA’s novel enzymatic DNA synthesis (EDS) technology for potential application by Pfizer for the company’s mRNA-based vaccines and other biopharma products. Codex DNA is a leader in developing automated solutions for on-demand synthesis of genes and mRNA.
The financial terms of the accord include an upfront payment from Pfizer to Codex DNA, along with success-based technical milestone payments that could be earned during the near term. Codex DNA is additionally eligible to receive additional milestone payments based on the achievement of specified development, regulatory, and commercialization goals associated with any products developed from the application of Codex DNA’s technology developed and licensed under the deal.
“We believe this strategic, early access collaboration and licensing arrangement is a validation of our cutting-edge SOLA enzymatic DNA synthesis technology and has the potential to accelerate vaccine and biotherapeutic research and development programs for the benefit of humanity,” stated Todd R. Nelson, Ph.D., CEO of Codex DNA.
BioNTech: 21st century immunotherapy powerhouse
Biopharmaceutical New Technologies (BioNTech) is a next-generation immunotherapy company pioneering novel therapies for cancer and other serious diseases. BioNTech exploits a wide range of computational discovery and therapeutic drug platforms for the rapid development of novel biopharmaceuticals. The company’s broad portfolio of oncology product candidates includes individualized and off-the-shelf mRNA-based therapies, innovative chimeric antigen receptor T cells, bispecific checkpoint immuno-modulators, targeted cancer antibodies and small molecules. Based on a deep expertise in mRNA vaccine development and in-house manufacturing capabilities, BioNTech and the company’s collaborators are developing multiple mRNA vaccine candidates for an array of infectious diseases alongside a diverse oncology pipeline. BioNTech has established a broad set of relationships with multiple worldwide pharma collaborators including Genmab, Sanofi, Bayer Animal Health, Genentech, Regeneron, Genevant, Fosun Pharma, and of course Pfizer.
BioNTech is advancing a deep and extensive portfolio of product candidates from four drug classes concentrated on treating cancer, infectious and rare diseases.
BioNTech and InstaDeep agreed in January 2022 on the development of a new computational method that analyses worldwide available sequencing data and predicts high-risk variants of SARS-CoV-2. The Early Warning System (EWS) developed in collaboration by the two companies is based on artificial intelligence (AI) calculated immune escape and fitness metrics.
The new method combines structural modeling of the viral Spike protein and AI algorithms to quickly flag potential high-risk variants entered into SARS-CoV-2 sequence data repositories within less than one day based on metrics scoring their fitness (ie; ACE2 and variant Spike protein interaction) as well as their immune escape properties. BioNTech and InstaDeep validated these predictions using experimental data generated in-house and publicly available information.
According to the companies, the results from the study underline that the EWS is capable of assessing new variants in minutes and risk monitoring variant lineages nearly in real-time. The EWS is additionally fully scalable as new variant data becomes available.
“With the advanced computational methods we have been developing over the past months we can analyze sequence information of the Spike protein and rank new variants according to their predicted immune escape and ACE2 binding score,” stated Ugur Sahin, M.D., CEO and Co-Founder of BioNTech. “Early flagging of potential high-risk variants could be an effective tool to alert researchers, vaccine developers, health authorities and policy makers, thereby providing more time to respond to new variants of concern.”
The company’s FixVac product candidates are composed of selected combinations of unmodified, pharmacologically optimized mRNA, encoding known cancer-specific shared antigens. The FixVac product candidates feature BioNTech’s proprietary immunogenic mRNA backbone and proprietary RNA-lipoplex, or RNA-LPX, delivery formulation, designed to enhance stability and translation, target dendritic cells and trigger innate and adaptive immune responses. The company is assessing FixVac product candidates in clinical studies, including the leading candidate BNT111 in a Phase II trial in patients with anti-PD-1-refractory/relapsed unresectable Stage III or IV melanoma. The investigational cancer immunotherapy was granted Fast Track Designation by the FDA in November 2021. BNT113 has advanced into a Phase II study in HPV16-positive head and neck cancers.
BioNTech is a pioneer and worldwide leader in developing fully individualized cancer immunotherapies. The company has developed a first-of-its-kind, on-demand manufacturing process to treat every individual patient based on the mutation profile of the patient’s tumor, and this treatment approach has been validated in the clinic in collaboration with Roche Group member Genentech.
BioNTech’s proprietary individualized mRNA-based cancer vaccine platform iNeST (Individualized Neoantigen Specific Immunotherapy) also has advanced a product candidate into Phase II development. On October 1, 2021, the company announced that the first colorectal cancer patient was treated with the individualized mRNA cancer vaccine BNT122 (autogene cevumeran, RO7198457) in a Phase II study. The trial was initiated in the United States, Germany, Spain and Belgium. BioNTech is jointly developing BNT122 with Genentech in other trials.
Autogene cevumeran is the lead candidate from BioNTech’s mRNA-based cancer vaccine platform. Since 2016, the company has advanced mRNA-based cancer vaccines targeting neoantigens in collaboration with Genentech, including the joint clinical development of autogene cevumeran in a Phase Ia/Ib basket study in solid tumors and a randomized Phase II trial in first-line melanoma patients, which was initiated during 2019. BioNTech and Genentech are equally sharing development costs and potential profits from their joint development of mRNA-based cancer vaccines targeting neoantigens for the potential treatment of various cancers.
BioNTech and Crescendo Biologics – a clinical-stage immuno-oncology company developing novel, targeted T cell enhancing therapeutics – announced in January 2022 a multi-target discovery collaboration to develop novel immunotherapies for treating patients with cancer and other diseases. According to the companies, the initial term of the discovery collaboration is three years.
The collaboration leverages BioNTech’s proprietary multimodal immunotherapy expertise with Crescendo’s proprietary Humabody VH platform to develop precision immunotherapies. These include mRNA-based antibodies and engineered cell therapies against targets selected by BioNTech, which will hold exclusive global development and commercialization rights to all immunotherapies stemming from the collaboration.
MODERNA
In the decade since inception, Moderna has transformed from a research-stage company advancing programs in the field of messenger RNA, to an enterprise with a diverse clinical portfolio of vaccines and therapeutics across seven modalities, a broad intellectual property portfolio in fields including mRNA and lipid nanoparticle formulation, and an integrated manufacturing plant that allows for clinical and commercial production at scale and at unprecedented speed. The company’s capabilities have united to allow the authorized use of one of the earliest and most-effective vaccines against the COVID-19 pandemic.
Moderna’s messenger RNA platform builds on continuous advances in basic and applied mRNA science, delivery technology and manufacturing, and has enabled the development of therapeutics and vaccines for infectious diseases, immuno-oncology, rare diseases, cardiovascular diseases, and auto-immune diseases.
Moderna’s mission is to deliver on the promise of messenger RNA science to create a new generation of transformative medicines for patients. Moderna during 2019 announced dosing of the first monoclonal antibody encoded by mRNA in a clinical trial.
Moderna’s science formed the backbone of the development program for the biotechnology company’s original COVID-19 vaccine, Spikevax (mRNA-1273), which was its first product to market.
Moderna reported that 807 million doses of the company’s COVID-19 vaccine shipped globally during 2021, with 25 percent of those doses shipped to low-income and middle-income countries. The more than 200 million doses delivered to low-income and middle-income countries in 2021 were more than the company delivered to any other country or multinational group last year, other than the United States.
Moderna continues to scale, with 40 programs in development including 23 in clinical studies covering mRNA infectious disease vaccines and mRNA therapeutics spanning seven different modalities. The company and collaborators have published almost 100 peer-reviewed manuscripts.
According to CEO Stéphane Bancel, “We will continue to advance mRNA vaccines that can have a profound impact on health and quality of life including vaccines against respiratory viruses with the goal of bringing to market a pan-respiratory annual customizable booster vaccine. In parallel, we are advancing first-in-class vaccines against latent viruses, which remain in the body for life and can cause lifelong medical conditions and we are also working to bring to market therapeutics based on mRNA-encoded proteins to help address multiple disease areas. We look forward to further leveraging our mRNA technology and delivery into gene-editing and other ways to impact human health.”
Moderna has been developing new versions of mRNA-1273 to combat variants of the novel coronavirus, including:
• mRNA-1273.351 for Beta variant
• mRNA-1273.617 for Delta variant
• mRNA-1273.211 for Beta variant + wild-type
• mRNA-1273.213 for Beta+Delta variant
• mRNA-1273.529 for Omicron variant
• mRNA-1283 for next generation (2-5 ºC)
Moderna announced on January 26 that the first participant was dosed in the Phase II trial of the company’s Omicron-specific booster candidate, mRNA-1273.529. Moderna also announced the publication of neutralizing antibody data against the Omicron variant six months following a booster dose in The New England Journal of Medicine. While Omicron neutralization had decreased 6.3-fold from peak titers at day 29 post-boost, levels remained detectable in all participants. Neutralizing titers against Omicron decreased more rapidly than titers versus the ancestral strain of the virus (D614G), which declined 2.3-fold over the same time frame.
Moderna’s industry-leading mRNA pipeline extends well beyond the world of COVID. For example, Moderna is developing adult vaccines for flu (mRNA-1030), COVID + flu (mRNA-1073), and respiratory syncytial virus (mRNA-1345).
The first participants were dosed in the Phase II/III trial of mRNA-1345, known as ConquerRSV. The primary purpose of the study’s Phase II segment is to test the safety of the mRNA-1345 vaccine in adults older than 60 years of age for initiation of the large‑scale Phase III segment. The primary purpose of the Phase III segment is to establish the safety and efficacy of mRNA-1345 in adults older than 60 years of age in support of licensure. Moderna plans to enroll 34,000 participants into the study. There is no vaccine approved for the prevention of RSV.
The company’s mRNA vaccine portfolio includes candidates to combat respiratory viruses, tropical viruses, and latent viruses. Moderna is dedicated to developing first-in-class vaccines against latent viruses for which there are no approved vaccines, including vaccines against Epstein-Barr Virus (EBV), cytomegalovirus (CMV) and human immunodeficiency virus (HIV).
Moderna is developing the HIV vaccines mRNA-1644 and mRNA-1574.
On January 27, 2022, Moderna and the nonprofit scientific research organization IAVI announced that first doses were administered in a clinical trial of experimental HIV vaccine antigens at George Washington University (GWU) School of Medicine and Health Sciences in Washington, D.C. The Phase I study, funded by the Bill & Melinda Gates Foundation, is designed to evaluate the hypothesis that sequential administration of priming and boosting HIV immunogens delivered by mRNA can induce specific classes of B-cell responses and guide their early maturation toward broadly neutralizing antibody (bnAb) development. Moderna says the induction of bnAbs is widely considered to be a goal of HIV vaccination, and this is the first step in that process. The immunogens being assessed in IAVI G002 were developed by scientific teams at IAVI and Scripps Research and will be delivered via Moderna’s mRNA technology.
On January 5, the company announced the first participant had been dosed in the Phase I trial (Eclipse) of Moderna’s EBV vaccine candidate. Moderna’s mRNA-1189 is being developed to prevent EBV-induced infectious mononucleosis (IM) and potentially EBV infection. Similar to Moderna’s CMV vaccine candidate mRNA-1647, mRNA-1189 contains four mRNAs that encode EBV envelope glycoproteins (gH, gL, gp42, gp220), which mediate viral entry into B-cells (a type of immune system cells) and epithelial surface cells, the major targets of EBV infection. There is no approved vaccine for EBV or IM. Moderna says potential future indications may be the prevention of EBV reactivation in other types of conditions including post-transplant lymphoproliferative disease.
According to Moderna, the seasonal flu vaccine candidate mRNA-1010 successfully boosted hemagglutination inhibition (HAI) assay geometric mean titers against all strains 29 days after vaccination at all doses tested in younger and older adults in a Phase I study and no significant safety concerns were observed. A Phase II trial of mRNA-1010 is fully enrolled, and preparation for Phase III development is underway. mRNA-1010 encodes for hemagglutinin (HA) glycoproteins of four flu strains and targets lineages recommended by the World Health Organization for flu prevention, including seasonal influenza A H1N1, H3N2 and influenza B Yamagata and Victoria.
Moderna announced on December 10, 2021, two development candidates which the company believes may expand coverage against seasonal influenza strains. mRNA-1011 will have one additional HA antigen and mRNA-1012 will have two additional HA antigens. Moderna is additionally developing two next-generation flu candidates that incorporate neuraminidase antigens to potentially improve immunity by elevating immunologic breadth targeting more conserved antigens (mRNA-1020 and mRNA-1030).
The first participants were dosed in a Phase III trial of the CMV vaccine candidate mRNA-1647. Known as CMVictory, the trial is investigating the safety and efficacy of mRNA-1647 against primary CMV infection in women ages 16-40 years. Moderna intends to enroll up to 8,000 participants in the study, including 6,900 women of child-bearing age, at 150 sites around the world, beginning in the United States. The company has set a goal of enrolling a diverse group of U.S. participants, including 42 percent of participants who are Persons of Color. Based on the interim analysis of the Phase II trial, the 100 μg dose was selected for the Phase III pivotal trial.
On the therapeutics front, recent pipeline progress has included a Phase II trial (EPICCURE) of the mRNA therapeutic AZD8601 that encodes for vascular endothelial growth factor-A (VEGF-A) in patients undergoing coronary artery bypass grafting (CABG). The study’s primary endpoint of safety and tolerability was met. Moderna has licensed global commercial rights to AZD8601 to AstraZeneca.
Initial data from the Phase 1 study of OX40L/IL-23/IL-36γ (Triplet) (mRNA-2752) in patients with accessible solid tumors and lymphoma demonstrated that mRNA-2752 given in combination with AstraZeneca’s durvalumab (marketed as Imfinzi) was tolerated at all dose levels tested and elicited evidence of anti-tumor activity.
Moderna and Carisma Therapeutics, a biopharmaceutical pioneer in engineered macrophage-based therapeutics, entered into a strategic collaboration deal during January to discover, develop and commercialize in vivo engineered chimeric antigen receptor monocyte (CAR-M) therapeutics for treating cancer, including solid tumors. The collaboration combines Carisma’s engineered macrophage technology with Moderna’s mRNA and LNP technologies to generate and develop in vivo CAR-M therapeutics. The multi-year research collaboration is being funded by Moderna with options for up to 12 targets for development and commercialization.
Moderna is investing in a new science center, known as the Moderna Science Center based in Cambridge, Mass. The 462,000 square foot state-of-the-art building is targeting LEED Zero certification and is being designed to be the most sustainable commercial lab building located in Cambridge.
Moderna launched the company’s Artificial Intelligence (AI) Academy in partnership with Carnegie Mellon University. The AI Academy is intended to educate and empower company employees to integrate AI and machine learning solutions into their efforts to advance mRNA medicines.
Johnson & Johnson/Janssen
The Janssen COVID-19 Vaccine is authorized for use in the United States under an Emergency Use Authorization for active immunization to prevent Coronavirus Disease 2019 caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). On October 20, 2021, the U.S. Food and Drug Administration issued an EUA for a booster dose of the company’s vaccine for adults aged 18 and older at least two months following primary vaccination with single-shot Johnson & Johnson COVID-19 vaccine; and for eligible individuals who received a different authorized or approved COVID-19 vaccine. J&J’s booster shot is the same formulation and dosage as the primary shot.
A single booster dose of J&J’s COVID-19 vaccine may additionally be administered as a heterologous booster dose following completion of primary vaccination with another authorized or approved COVID-19 vaccine. J&J says the eligible population(s) and dosing interval for the heterologous booster dose are the same as those authorized for a booster dose of the vaccine used for primary vaccination.
“Our data support a schedule that provides benefit to individuals based on their risks associated with COVID-19, whether administered as a single dose for an efficient response to the pandemic, or as a booster dose after at least two months – to protect against symptomatic COVID-19,” stated Paul Stoffels, M.D., Vice Chairman of the Executive Committee and Chief Scientific Officer, Johnson & Johnson. “We also welcome the FDA’s decision to include a heterologous boosting option as part of this authorization. The ability to boost immune responses regardless of the primary vaccine regimen an individual has received provides more flexibility in protecting those already immunized, and is very beneficial to global public health as we look to curb this pandemic.”
On January 6, 2022, J&J unveiled new results from the largest study to date on the durability of COVID-19 vaccines in the United States, demonstrating that a single shot of the Johnson & Johnson vaccine resulted in long-lasting protection for up to six months against COVID-19 breakthrough infections, hospitalizations, and intensive care unit (ICU) admissions. The study was sponsored by the Janssen Pharmaceutical Companies and performed in partnership with the Department of Science-Aetion, and the Division of Pharmacoepidemiology, Department of Medicine at Brigham and Women’s Hospital and Harvard Medical School.
The study posted on medRxiv comprehensively reviewed the durability profiles for all three vaccines authorized or approved in the United States using the same methodology across three outcomes of interest: COVID-19 breakthrough infections, hospitalizations, and ICU admissions. The study demonstrated that the effectiveness of J&J’s vaccine against breakthrough infections and hospitalizations remained durable. The mRNA vaccines (two-doses) demonstrated waning effectiveness for hospitalizations and breakthrough infections. All three vaccines showed no evidence of waning protection against COVID-19-related ICU admissions at any point, demonstrating strong sustained protection against critically severe disease. According to J&J, the study was not designed to compare the durability of vaccines.
Per J&J, comprehensive studies to date on the durability of all vaccines authorized or approved for use in the United States have been limited, with many concentrating on high-risk populations or specific geographic regions or states. This is the largest COVID-19 real-world effectiveness durability study in the United States and the first to analyze the durability of baseline protection up to six months for all three U.S. authorized or approved vaccines, and for three COVID-19 outcomes of interest (breakthrough infections, hospitalizations, and ICU admissions). Researchers used national claims, laboratory, and hospital data encompassing 168 million individuals to conduct a matched case-control study between January 1 and September 7, 2021 for 17 million fully vaccinated individuals matched on calendar time, 3-digit zip code, age, sex and comorbidity scores.
“We continue to undertake extensive efforts to study the durability of protection offered by the Johnson & Johnson vaccine amidst the ever-changing COVID-19 pandemic,” commented Mathai Mammen, M.D., Ph.D., Executive Vice President, Pharmaceuticals, Janssen Research & Development, Johnson & Johnson. “While these are rapidly evolving data, we are seeing vaccine effectiveness against COVID-19-related hospitalization of approximately 80 percent from a single shot of the Johnson & Johnson vaccine, and this level of protection holds steady across the length of time studied thus far – up to six months. The robust and sustained durability of our COVID-19 vaccine reflects its unique underlying immunology. We previously reported that our vaccine induces a strong antibody response as well as an especially strong increase in T-cells that is consistent across variants, including Omicron.”
J&J says these results add to the body of evidence showing that the Johnson & Johnson COVID-19 vaccine elicits protection against variants of concern. Preliminary data from the Phase IIIb Sisonke trial, carried out during November and December 2021, showed 85 percent effectiveness for the homologous (same vaccine) booster shot of the Johnson & Johnson vaccine, against COVID-19-related hospitalization in South Africa when Omicron was dominant.
J&J additionally announced during December that a heterologous booster (different vaccine) of the company’s vaccine in individuals who initially received the BNT162b2 mRNA vaccine generated a 41-fold increase in neutralizing antibody responses by four weeks following the boost and a 5-fold increase in CD8+ T-cells to Omicron by two weeks. A homologous boost with BNT162b2 generated a 17-fold increase in neutralizing antibodies by four weeks following the boost and a 1.4-fold increase in CD8+ T-cells by two weeks, according to J&J. The elevation in CD8+ T-cells generated by Johnson & Johnson’s vaccine may be key to explaining the high levels of effectiveness against severe COVID-19 disease and hospitalization.
In non-COVID-19 news from Janssen, the company on December 7, 2021, revealed additional vaccine efficacy and safety data from the Phase IIb CYPRESS trial of its respiratory syncytial virus (RSV) adult vaccine candidate. Results demonstrate that the vaccine candidate was highly effective in protecting against three clinical definitions of lower respiratory tract disease (LRTD) resulting from RSV, showing vaccine efficacy of 70 to 80 percent in adults aged 65 and older.
“Data from our Phase IIb CYPRESS study bring us another step closer to delivering a potentially first-in-class vaccine to help protect older adults from RSV,” remarked Penny Heaton, M.D., Global Therapeutic Area Head, Vaccines, Janssen Research & Development.
At the beginning of February 2022, the U.S. FDA approved Cabenuva (cabotegravir, rilpivirine) for use every two months, expanding the label of the first and only complete long-acting HIV treatment. Cabenuva is now approved for administration as few as six times annually for virologically suppressed adults living with HIV without prior treatment failure or resistance to cabotegravir or rilpivirine.
Cabenuva is composed of two separate injectable medicines, the non-nucleoside reverse transcriptase inhibitor (NNRTI) rilpivirine and the integrase strand transfer inhibitor (INSTI) cabotegravir. Rilpivirine is an extended-release injectable suspension in a single-dose vial and a product of Janssen Sciences Ireland Unlimited Company. Cabotegravir is an extended-release injectable suspension provided in a single dose vial.
Rilpivirine is approved in the United States as a 25-milligram tablet taken once daily for the treatment of HIV-1 in combination with other antiretroviral agents in antiretroviral treatment-naïve patients 12 years of age and older and weighing at least 35kg with a viral load ≤100,000 HIV RNA copies/ml.
“The expanded label approval for Cabenuva – to be administered every two months – marks an important step forward in advancing the treatment landscape for people living with HIV,” stated Candice Long, President, Infectious Diseases & Vaccines, Janssen Therapeutics, a Division of Janssen Products. “With this milestone, adults living with HIV have a treatment option that further reduces the frequency of medication.”
The Janssen R&D team continues to product new indications for already-marketed medicines, including the blockbuster brand Xarelto (rivaroxaban). During December 2021, the U.S. FDA approved two pediatric indications for Xarelto: the treatment of venous thromboembolism (VTE, or blood clots) and reduction in the risk of recurrent VTE in patients from birth to less than 18 years after at least five days of initial parenteral (injected or intravenous) anticoagulant treatment; and thromboprophylaxis (prevention of blood clots and blood-clot related events) in children aged 2 years and older with congenital heart disease who have undergone the Fontan procedure. Xarelto is the only direct oral anticoagulant (DOAC) to have received FDA clearance for primary prevention of clots in pediatric patients following the Fontan procedure and the first DOAC in the United States to offer an oral suspension formulation for flexible, body weight-adjusted dosing options for pediatric patients.
One of the world’s top-selling medicines, Xarelto has been approved in the United States for 11 indications – the most of any DOAC – and is the most studied oral Factor Xa inhibitor in its class. The December 2021 approval is based on two Phase III pediatric trials from the industry-leading EXPLORER clinical research program, EINSTEIN-Jr, the largest study to date assessing pediatric patients from birth to <18 years of age with previously diagnosed VTE; and UNIVERSE, the first clinical study to investigate a DOAC for the prevention of VTE in pediatric patients after recently undergoing the Fontan procedure.
In other J&J blockbuster brand updates, new data were reported from two studies investigating the efficacy and safety of Imbruvica (ibrutinib) plus venetoclax (sold under the brand names Venclexta and Venclyxto) as a potential fixed-duration treatment in adults with previously untreated chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL). New secondary endpoint data from the Phase III GLOW trial (NCT03462719) demonstrated that fixed-duration treatment with I+V resulted in undetectable minimal residual disease (uMRD) responses that were deeper versus patients treated with chlorambucil plus obinutuzumab (Clb+O), and an additional analysis demonstrated that uMRD responses were better sustained during the first year post-treatment.
Imbruvica (ibrutinib) is a once-a-day oral medicine that is jointly developed and commercialized by Janssen Biotech and Pharmacyclics, an AbbVie company. The medication blocks the Bruton’s tyrosine kinase (BTK) protein, which is needed by normal and abnormal B cells, including specific cancer cells, to multiply and spread. By blocking BTK, research shows that Imbruvica may help move abnormal B cells out of their nourishing environments and inhibits their proliferation.
New Tremfya (guselkumab) efficacy and safety data were reported by Janssen from the Phase IIIb COSMOS study published in Annals of the Rheumatic Diseases (ARD), assessing the selective interleukin (IL)-23 inhibitor in adults with active psoriatic arthritis (PsA) who showed inadequate efficacy or intolerance to tumor necrosis factor inhibition (TNFi). Results demonstrated significantly higher proportions of patients treated with Tremfya had improvement in joint signs and symptoms and complete skin clearance compared to placebo at week 24 in this documented TNFi-IRa patient population, which is often more difficult to treat. Additionally, improvements in signs and symptoms of PsA were maintained or numerically increased through one year (week 48) among Tremfya-randomized patients.
Developed by Janssen, Tremfya is the first approved fully human monoclonal antibody that selectively binds to the p19 subunit of IL-23 and inhibits its interaction with the IL-23 receptor. IL-23 is a significant driver of the pathogenesis of inflammatory diseases, including moderate-to-severe plaque PsO and active PsA.
The FDA approved Darzalex Faspro (daratumumab and hyaluronidase-fihj) in combination with Kyprolis (carfilzomib) and dexamethasone for patients with multiple myeloma after first or subsequent relapse. Announced on December 1, 2021, the marketing clearance is the ninth indication for Darzalex Faspro, the only subcutaneous anti-CD38 monoclonal antibody approved across a range of standard treatment regimens.
“Today’s approval of Darzalex Faspro, in combination with yet another widely used regimen, further substantiates the subcutaneous formulation as a foundational element in the treatment of multiple myeloma,” said Craig Tendler, M.D., global head of Late Development, Diagnostics & Medical Affairs, Hematology & Oncology, Janssen Research & Development.
New analyses were reported by Janssen illustrating responses that first-line treatment with Darzalex (daratumumab)-based regimens may be able to achieve, including a potential survival benefit for the medication in combination with lenalidomide and dexamethasone (Rd). Updated data from the randomized Phase II GRIFFIN trial in transplant-eligible patients and real-world evidence in transplant-ineligible patients were presented at the ASH 2021 Annual Meeting. The data demonstrate improved outcomes with the addition of Darzalex to bortezomib (Velcade), lenalidomide (Revlimid) and dexamethasone (VRd), followed by Darzalex plus lenalidomide (R) maintenance therapy, in transplant-eligible patients.
Janssen Pharmaceutical announced near the end of December 2021 the submission of a Biologics License Application (BLA) to the U.S. FDA seeking approval of teclistamab for treating patients with relapsed or refractory (R/R) multiple myeloma (MM). Teclistamab is an investigational, off-the-shelf, T-cell redirecting, bispecific antibody that targets B-cell maturation antigen (BCMA) and CD3.
The BLA submission for teclistamab is supported by clinical data from MajesTEC-1 (NCT04557098, NCT03145181), an open-label, multicenter study assessing the safety and efficacy of teclistamab in adults with R/R multiple myeloma. In the clinical trial, investigators tested efficacy outcomes, including overall response rate, very good partial response and complete response using International Myeloma Working Group (IMWG) criteria, as well as the safety profile of teclistamab. Updated MajesTEC-1 data were presented at the December 2021 American Society of Hematology annual meeting.
“Despite all the gains that have been made in treating multiple myeloma, the unmet need still remains very high. Our relentless pursuit of treatments for this disease continues with the same sense of urgency that we have always had,” stated Peter Lebowitz, M.D., Ph.D., Global Therapeutic Area Head, Oncology, Janssen Research & Development. According to Dr. Mammen, “The deep expertise, creativity and persistence of the entire Janssen R&D organization enabled the expeditious advancement of teclistamab for multiple myeloma.”
In other December news on the multiple myeloma front, Janssen reported results from the Phase Ib/II CARTITUDE-1 trial testing the efficacy and safety of ciltacabtagene autoleucel (cilta-cel) in treating patients with relapsed and/or refractory MM. The investigational B-cell maturation antigen (BCMA)-directed chimeric antigen receptor T-cell (CAR-T) therapy is administered as a single infusion. The data, featured as an oral presentation at the American Society of Hematology (ASH) 2021 Annual Meeting and selected as part of the Highlights of ASH program, demonstrate that patients receiving cilta-cel continue to show deep and durable responses, with a very high overall response rate (ORR) of 98 percent.
Updated results were reported by Janssen from the MonumenTAL-1 Phase I first-in-human dose-escalation study of talquetamab (NCT03399799). Talquetamab is the first investigational off-the-shelf T cell redirecting bispecific antibody undergoing clinical development targeting both GPRC5D, a novel multiple myeloma target, and CD3 on T cells. Study results demonstrate that no new safety signals were observed with longer follow-up.
Janssen is developing new indications for niraparib, an orally administered, poly-ADP ribose polymerase (PARP) inhibitor being evaluated for treating patients with prostate cancer. Ongoing studies include the Phase III AMPLITUDE trial assessing niraparib in combination with abiraterone acetate plus prednisone in a biomarker-selected patient population with metastatic castration-resistant prostate cancer (mCSPC); the Phase III MAGNITUDE trial investigating niraparib in combination with abiraterone acetate plus prednisone as a first-line treatment option versus abiraterone acetate and prednisone plus placebo in adults with mCRPC; and QUEST, a Phase Ib/II study of niraparib combination therapies for treating mCRPC.
During April 2016, Janssen Biotech entered a global (except Japan) collaboration and license deal with Tesaro (acquired by GlaxoSmithKline in 2018), for exclusive rights to niraparib in prostate cancer. Niraparib is marketed by GlaxoSmithKline as Zejula for a variety of non-prostate cancer indications.
2022: Updates For Other Leading COVID-19 Players
Merck and Ridgeback Biotherapeutics announced on January 28 data from six preclinical studies showing that molnupiravir, an investigational oral antiviral COVID-19 medicine, was active against the SARS-CoV-2 variant Omicron (B1.1.529) in vitro. “These findings from multiple independent in vitro studies showing that molnupiravir has consistent antiviral activity against Omicron, the primary variant circulating globally, provide additional confidence in the potential of molnupiravir as an important treatment option for certain adults with mild to moderate COVID-19 who are at high risk for progressing to severe disease,” stated Dr. Dean Y. Li, President, Merck Research Laboratories. Merck is developing molnupiravir in collaboration with Ridgeback Biotherapeutics, a Miami-based biotech company. The investigational, orally administered nucleoside analog has been authorized for use in more than 10 countries, including in the United States, United Kingdom and Japan.
By early February, Merck had shipped more than 4 million courses of therapy to more than 25 countries, including 3 million courses to the U.S. government as part of a procurement agreement. Merck and Ridgeback are engaged in various efforts to accelerate broad, equitable access worldwide, including a recent deal on the allocation of up to 3 million courses of therapy to the United Nations Children’s Fund (UNICEF) for use in adults.
Molnupiravir’s 2021 sales of $952 million were all accounted for during the fourth quarter, mainly consisting of sales in the United States, the U.K. and Japan. On February 3, Merck projected that molnupiravir would generate sales of $5 billion to $6 billion in 2022, with the company producing 30 million treatment courses during the year.
Novavax, a biotechnology company dedicated to developing and commercializing next-generation vaccines for serious infectious diseases, announced on January 31 that it had filed a request to the U.S. FDA for an EUA for NVX-CoV2373. The protein-based COVID-19 vaccine candidate is intended for immunization of individuals 18 year of age and older against SARS-CoV-2. NVX-CoV2373 has been granted conditional authorization by multiple regulatory agencies around the globe, including the European Commission, and Emergency Use Listing (EUL) from the World Health Organization (WHO), with additional filings under review.
“We believe our vaccine offers a differentiated option built on a well-understood protein-based vaccine platform that can be an alternative to the portfolio of available vaccines to help fight the COVID-19 pandemic,” stated Novavax President and Chief Executive Officer Stanley C. Erck.
GlaxoSmithKline and Vir Biotechnology reported on January 11 a U.S. government deal to purchase additional supply of sotrovimab, authorized for the early treatment of COVID-19. The 600,000 additional doses were to be supplied to the U.S. government for distribution during first-quarter 2022, bringing the total amount of doses secured to date through binding agreements to 1.7 million worldwide.
Preclinical data generated via pseudo-virus and live virus testing show sotrovimab retains activity against all tested SARS-CoV-2 variants of concern, including Delta and Omicron. Sotrovimab was granted an EUA by the U.S. FDA during May 2021 as an investigational single-dose intravenous (IV) infusion SARS-CoV-2 monoclonal antibody.
GSK is collaborating with several organizations on COVID-19 vaccines by providing access to the company’s adjuvant technology. GSK is working with Sanofi, Medicago and SK bioscience to develop adjuvanted, protein-based vaccine candidates, all of which are undergoing Phase III development. GSK and Sanofi announced during December 2021 positive preliminary booster data for their COVID-19 vaccine candidate and continuation of a Phase III study per an independent Monitoring Board recommendation. Positive booster data demonstrate that neutralizing antibodies increased across all primary vaccines received (mRNA or adenovirus) in a 9- to 43-fold range and for all age groups tested, with a good safety and tolerability profile.
Gilead Sciences reported that Veklury sales increased 98 percent to $5.6 billion for full-year 2021 compared to the previous calendar term. Gilead received U.S. approval on January 21, 2022, to expand the Veklury indication to include the treatment of non-hospitalized adult and adolescent patients who are at high risk of progression to severe COVID-19, including hospitalization or death. The pediatric EUA was additionally expanded to include use of Veklury in non-hospitalized pediatric patients weighing at least 3.5 kg who are younger than 12 years of age or weigh less than 40 kg and who are at high risk of disease progression.
“Remdesivir has now helped to treat more than 10 million people around the world with COVID-19 and continues to play a key role in helping to reduce the burden of the pandemic. Based on the most recent data, we now understand that remdesivir is also effective in the early stages of COVID-19 infection, in addition to helping patients who are hospitalized with the disease,” remarked Gilead Sciences Chairman and Chief Executive Officer Daniel O’Day. “While we continue to advance remdesivir to benefit more patients in multiple settings, we are also advancing our investigational oral compounds. These are based on the same antiviral mechanism of action as remdesivir and a Phase I trial for our oral COVID-19 antiviral, GS-5245, is now underway.”
The U.S. FDA revised the authorizations for two monoclonal antibody treatments – bamlanivimab and etesevimab (administered together) and REGEN-COV (casirivimab and imdevimab) – to limit their use to only when a patient is likely to have been infected with or exposed to a variant that is susceptible to these treatments. FDA officials said because data demonstrate these treatments from Lilly and Regeneron Pharmaceuticals are highly unlikely to be active against the Omicron variant, which has been circulating at a very high frequency throughout the United States, these treatments are not authorized for use in any U.S. states, territories and jurisdictions at this time.
AstraZeneca reported on January 13 positive results from a preliminary analysis of a continuing safety and immunogenicity study (D7220C00001) demonstrating that Vaxzevria (ChAdOx1-S [Recombinant]), when given as a third dose booster, increased the immune response to Beta, Delta, Alpha and Gamma SARS-CoV-2 variants. A separate analysis of samples from the study demonstrated increased antibody response to the Omicron variant. A separate Phase IV study reported in a preprint with The Lancet on SSRN showed that a third dose of Vaxzevria substantially increased antibody levels following a primary vaccine series with Sinovac Biotech’s CoronaVac. AstraZeneca says these data add to the growing body of evidence supporting Vaxzevria as a third dose booster irrespective of the primary vaccination schedules tested.
Vaxzevria was invented by the University of Oxford, and is manufactured and supplied by the Serum Institute of India under the name Covishield via a sub-license deal. The vaccine has been granted a conditional marketing authorization or emergency use in more than 90 countries, and has received Emergency Use Listing from the World Health Organization.
Valneve announced on January 25 the start of booster vaccinations in adults from the French specialty vaccine company’s Phase III pivotal study, Cov-Compare. This booster extension is intended to provide homologous and first heterologous booster data to complement previous positive Phase I/II booster results. The study extension is assessing a booster dose of VLA2001 in adults, aged 18 years and older, who received primary vaccination with two doses of VLA2001, as well as participants, aged 30 and older, who received two doses of AstraZeneca’s AZD1222.
In other news reported on January 19, preliminary laboratory studies show that three doses of VLA2001 induced neutralization of the Omicron variant (B.1.1.529 lineage). Valneve commenced rolling submissions for initial approval of VLA2001 with the European Medicines Agency, the UK MHRA and the Bahraini NHRA following positive Phase III results. Valneva anticipates potential regulatory approvals during first-quarter 2022.
Clover Biopharmaceuticals, a global clinical-stage biotech company developing novel vaccines and biologic therapeutic candidates, announced on January 5 that the first participants were dosed with Clover’s COVID-19 vaccine candidate, SCB-2019 (CpG 1018/Alum), as a homologous booster dose following primary vaccination of SCB-2019 (CpG 1018/Alum) in the continuing worldwide Phase II/III SPECTRA study. Clover reported final efficacy data for SCB-2019 in SPECTRA during in 2021 and the clinical trial is continuing to generate additional immunogenicity and safety data.
A unique comparative study conducted at the Italian Spallanzani Institute – the leading Italian research institute for infectious diseases – by a joint Italian-Russian team of researchers representing the institute and the Gamaleya Center showed that the Sputnik V coronavirus vaccine demonstrates more than two times higher titers of virus neutralizing antibodies to the Omicron (B.1.1.529) variant than two doses of the Pfizer vaccine (2.1 times higher in total and 2.6 times higher 3 months after vaccination). Previous studies also showed additional significant strengthening of protection against Omicron by the Sputnik Light booster, which can additionally be a universal booster to other vaccines to strengthen and lengthen their protection against Omicron. Sputnik V has been authorized in 71 countries with a total population of more than 4 billion people, and Sputnik Light in 30-plus countries.
Bharat Biotech, a worldwide leader in vaccine innovation and developer of vaccines for infectious diseases, unveiled results on January 12 from a study conducted at Emory University showing that sera from subjects who received a booster dose of Covaxin (BBV152) six months after getting a primary two-dose series of Covaxin, neutralized the SARS-CoV-2 Omicron and Delta variants. Earlier studies showed the neutralizing potential of Covaxin against SARS-CoV-2 Variants of Concern Alpha, Beta, Delta, Zeta and Kappa.
Developed in collaboration with the Indian Council of Medical Research (ICMR) – National Institute of Virology, Covaxin is a highly purified and inactivated vaccine manufactured using a vero cell manufacturing platform. With more than 200 million doses having been administered to adults and children outside the United States, the vaccine is authorized under emergency use in more than 20 countries, and an EUA is in process in more than 60 other countries.
CanSino Biologics announced on January 11 that Preprints with The Lancet, a collaboration between the research-sharing platform SSRN and The Lancet, published a clinical trial on the safety and immunogenicity of CanSinoBIO’s Recombinant COVID-19 Vaccine (Adenovirus Type 5 Vector) for Inhalation Convidecia as a heterologous booster. The study results indicated that a heterologous booster with one dose of Inhalation Convidecia for adults aged 18 years and older, who have received two doses of inactivated COVID-19 vaccine, can induce a higher level of neutralizing antibodies than those with a homogeneous booster of inactivated vaccine.
A study demonstrated that the protein-based COVID-19 vaccine NVSI-06-07 manufactured by Sinopharm, when given as a booster after two doses of an earlier shot from the Chinese firm, elicited a stronger antibody response against the Omicron variant than a third dose of the original. The study – which was published on January 4 and had not been peer reviewed – arose from concerns over the effectiveness of Sinopharm’s BBIBP-CorV jab, one of the two leading COVID-19 vaccines exported by China, against the Omicron variant.
An earlier study demonstrated a BBIBP-CorV booster generated weaker neutralization against Omicron than versus an older coronavirus strain from the central Chinese city of Wuhan. NVSI-06-07 was approved for emergency use as a booster in the United Arab Emirates in December 2021 and incorporates a different technology than BBIBP-CorV, which contains an inactivated form of the coronavirus.
Celltrion Group announced on January 3 results for the company’s cocktail therapy candidates, including neutralization data against the Omicron variant. The Phase I study is a randomized, double-blind and placebo-controlled trial designed to investigate the safety, tolerability and pharmacokinetics of regdanvimab (product code CT-P63) in 24 healthy subjects in Poland. The clinical trial met its primary objectives with data demonstrating regdanvimab was safe and well tolerated, with no significant drug-related adverse events. The European Commission granted marketing authorization for regdanvimab following a positive opinion issued by the European Medicines Agency’s Committee for Medicinal Products for Human Use during November 2021.
Zydus Group announced on February 2 that the company started supplies of its COVID-19 vaccine ZyCoV-D to the Government of India against their order from its newly commissioned state-of-the-art, Zydus Vaccine Technology Excellence Centre at the Zydus Biotech Park in Changodar, Ahmedabad. The group is additionally planning to make the vaccine available in the private market. ZyCoV-D is a three-dose vaccine administered intradermally using the painless PharmaJet needle free system, Tropis, on day 0, day 28 and day 56.



")

