
Novartis 2017: Back To The Future
2017 is serving as another challenging year for Novartis as the company continues to work through the Gleevec/Glivec patent expirations that began in 2016, but management is confident that a new phase of growth will start in 2018.
![]()
Novartis AG
Lichtstrasse 35
4056 Basel, Switzerland
Telephone: +41 61 324 11 11
Website: novartis.com
Best-Selling Products
| Product | 2016 Sales | 2015 Sales |
|---|---|---|
| Gleevec/Glivec | $3,323 | $4,658 |
| Gilenya | $3,109 | $2,776 |
| Lucentis | $1,835 | $2,060 |
| Tasigna | $1,739 | $1,632 |
| Sandostatin | $1,646 | $1,630 |
| Afinitor/Votubia | $1,516 | $1,607 |
| Galvus, Eucreas | $1,193 | $1,140 |
| Cosentyx | $1,128 | $261 |
| Diovan, Diovan HCT | $1,073 | $1,284 |
| Exjade | $956 | $917 |
| Exforge | $926 | $1,047 |
| Xolair | $835 | $755 |
| Votrient | $729 | $565 |
| Tafinlar/Mekinist | $672 | $453 |
| Promacta/Revolade | $635 | $402 |
| Travoprost Group | $619 | $631 |
| Jakavi | $581 | $410 |
| Voltaren/Cataflam | $525 | $558 |
| Neoral, Sandimmun | $515 | $570 |
All sales are in millions of dollars.
Financial Performance
| 2016 | 2015 | |
|---|---|---|
| Revenue | $48,518 | $49,414 |
| Net income | $6,698 | $17,794 |
| Diluted EPS | $2.80 | $7.29 |
| R&D expense | $9,039 | $8,935 |
| 1H2017 | 1H2016 | |
|---|---|---|
| Revenue | $23,781 | $24,070 |
| Net income | $3,644 | $3,817 |
| Diluted EPS | $1.54 | $1.60 |
| R&D expense | $4,231 | $4,231 |
All sales are in millions of dollars except EPS.
Novartis confronted several pressing issues during 2016, including the loss of U.S. patent protection for the cancer therapy Gleevec, returning the company’s eye care division to growth, and accelerating the uptake of the heart failure medicine Entresto. According to management, Novartis was able to maintain its sales momentum despite these challenges, though the company experienced a decrease in operating income.
Sales for Gleevec/Glivec (imatinib mesylate) decreased from $4.66 billion in 2015 to $3.32 billion during the 2016 calendar term. Despite the roughly $1.34 drop-off in sales year over year, the oncology medicine remained Novartis’ best-selling product in 2016. The sales decline continued during first-half 2017, falling to $1.05 billion.
Novartis’ Alcon is a worldwide leader in ophthalmology with product offerings in eye care devices and vision care. Alcon sales totaled $6 billion in 2015 and $5.81 billion during the following year. Sales were restated to reflect the new divisional structures and product transfers between divisions, announced on Jan. 27, 2016. Alcon’s first-half 2017 sales totaled $2.93 billion, which was the same amount as that generated during the first six months of 2016.
“One area where we fell short in 2016 was Alcon,” notes Novartis CEO Joseph Jimenez. “We started the year with the ambition of returning the business to growth. While we were successful in returning the Vision Care segment to growth in the second half, the Surgical business is taking longer than expected and is preventing a positive growth rate for the overall Alcon Division. We will continue to diligently execute the growth plan in 2017.”

“Novartis delivered very strong innovation in Q2 (2017) including the positive pivotal trial readouts for RTH258, ACZ885 and CTL019 JULIET, demonstrating the strength of our pipeline. We are on track for the full year guidance. The trajectory of the current growth drivers reinforces our confidence in our next growth phase, which we expect to start in 2018,” says Joseph Jimenez, CEO of Novartis.
In terms of the January 2016 announcement, Novartis at that time reported plans to further focus its divisions, integrating businesses that share therapeutic areas to better leverage the company’s development and marketing capabilities. Among the plans was the transfer of the Ophthalmic Pharmaceuticals franchise from the Alcon Division to the Innovative Medicines Division (formerly named the Pharmaceuticals Division), and the transfer of selected mature products from the Innovative Medicines Division to the Sandoz Division. Operationally, these transfers were completed effective April 1, 2016. The centralization of manufacturing and the integration of some drug development functions, additionally announced on Jan. 27, were operationally completed effective July 1, 2016.
“Last year we reshaped Novartis from a group of loosely affiliated divisions into an integrated company, consolidating several functions,” Jimenez stated. “We created a Global Drug Development organization to better share expertise, ensure optimal resource allocation, and leverage new technology platforms across divisions. At the same time, we created a single drug manufacturing organization that can better optimize production capacity and utilization, while taking steps to lower our costs.”
Entresto (sacubitril/valsartan) received U.S. and EU regulatory approval during 2015 for treating chronic heart failure. Entresto was launched in the United States during July 2015 and generated sales of $21 million during the rest of that year. The first-in-class angiotensin receptor/neprilysin inhibitor proceeded to produce 2016 global sales of $170 million, which was regarded as a disappointing amount in the analyst community that had projected a quick ascension to blockbuster sales for the product. First-half 2017 worldwide sales for Entresto reached $194 million.
“In 2016, Novartis continued to strengthen its business, accelerate innovation and further sharpen its organizational structure,” says Joerg Reinhardt, chairman of the board of directors for Novartis. “These steps are primarily designed to enhance our scientific and operating capabilities. … We are confident that our strategy of using science-based innovation to deliver better health outcomes for patients will reinforce our market position and increase sales, profits and shareholder value in the long term.”
As for 2017, first-half net sales were in line with the previous year’s total (0 percent constant currencies, -2 percent U.S. dollars), as the company’s growth drivers offset the Gleevec/Glivec Gx impact. Meanwhile, management has touted strong pipeline results during 2017 that underpin the potential of several highly innovative products and reinforce Novartis’ growth prospects.
“We have reshaped our company to build depth in areas where we are strongest and focus on innovation,” Jimenez comments. “We have a strong foundation for our next growth phase expected to start in 2018 with Cosentyx, Entresto, and other growth drivers such as Kisqali, as well as leading positions in the generics and eye care divisions.”
According to Novartis executives, the company is expanding partnerships with leading academic and private research institutes, with the goal of advancing developments in emerging frontiers, including gene editing and immuno-oncology. Management notes that partnerships are additionally vital for the company’ activities in digital health, as Novartis is working to improve evidence-based information about its products and to continue exploring pay-for-performance pricing models.
In early September, Jimenez announced his intent to step down as CEO in 2018, after eight years of serving in that post. Vasant (Vas) Narasimhan, M.D., global head of drug development and chief medical officer, will assume CEO duties effective Feb. 1, 2018. Dr. Narasimhan is a member of the executive committee and joined Novartis during 2005.
Mr. Jimenez states, “Both from a professional and a personal perspective, this is the right moment to hand the leadership reins of the company to Vas. Our strong pipeline and the strategic moves we have taken to focus the company have put Novartis on a strong path for the future.”
2017 Performance & Outlook
Based on the first-half 2017 results for Novartis, full-year net sales are expected to be broadly in line with 2016 at constant currencies (cc) after absorbing the impact of generic competition, including the continued genericization of Gleevec/Glivec in the United States and Europe. Core operating income is projected to be broadly in line or decline by low single digits (cc).
Management expects 2017 net sales performance (cc) for Innovative Medicines to be broadly in line with full-year 2016, to a slight increase; for Sandoz: broadly in line with the previous year; and for Alcon, guidance was revised upward to low single digit growth.
If mid-July exchange rates prevail for the remainder of 2017, the currency impact for the full year would be negative 1 percentage point on net sales and negative 2 percentage points on core operating income, according to Novartis.
The 2017 first-half net sales totaled $23.8 billion (-1 percent, +1 percent cc) as volume growth of 6 percentage points was partially offset by the negative impacts of generic competition (-3 percentage points) and pricing (-2 percentage points).
Operating income in first-half 2017 came in at $4.2 billion (-8 percent, -4 percent cc). Core adjustments amounted to $2 billion in line with first-half 2016, as the RLX030 net charge was offset mostly by lower amortization. RLX030 (serelaxin) is a novel recombinant form of the human hormone relaxin 2 undergoing late-stage studies, and is believed to act via multiple mechanisms to reduce stress on the heart, kidneys and other organs.
Core operating income was reported at $6.2 billion (-5 percent, -2 percent cc) for the 2017 first half. Core operating income margin in constant currencies declined 0.9 percentage points, mainly because of generic erosion of Gleevec/Glivec and growth investments. According to Novartis, currency had a negative impact of 0.2 percentage points, resulting in a net decrease of 1.1 percentage points to 26.3 percent of net sales.
Net income amounted to $3.6 billion (-5 percent, -1 percent cc), 4decreasing less than operating income due to higher income from associated companies. Earnings per share were $1.54 (-4 percent, -1 percent cc), including the benefit from the share buyback program.
Core net income came in at $5.6 billion (-3 percent, 0 percent cc), including the benefit from higher core income from associated companies. Core EPS was reported at $2.35 (-2 percent, +1 percent cc), including the benefit from the share buyback program.
Innovative Medicines net sales for the 2017 first half amounted to $16 billion (-1 percent, +2 percent cc), with volume growth of 7 percentage points driven by Cosentyx, Entresto, Promacta/Revolade, Jakavi, Tafinlar + Mekinist and Gilenya. Generic competition had a negative impact of 4 percentage points and pricing had a negative impact of 1 percentage point, largely because of Gleevec/Glivec genericization in Europe and the United States.
Sandoz net sales during the first two quarters of 2017 amounted to $4.9 billion (-3 percent, -2 percent cc), as volume growth of 6 percentage points was more than offset by 8 percentage points of price erosion. U.S. sales fell 8 percent (cc) compared to first-half 2016, mainly due to pricing pressure in retail generics. Net sales across Europe and the rest of the world rose 2 percent (cc) during January-June 2017. Novartis’ Sandoz Division is a worldwide leader in generic pharmaceuticals and biosimilars.
Alcon net sales came in at $2.9 billion (0 percent, +2 percent cc). First-half 2017 Surgical sales improved 1 percent (cc), fueled by strong performance of the vitreoretinal portfolio and cataract consumables. Vision Care sales increased 3 percent (cc) year over year, driven by the continued double-digit growth of Dailies Total1.
Underpinning Novartis’ financial results during the first six months of 2017 was a continued focus on key growth drivers, including Cosentyx, Entresto, Promacta/Revolade, Tafinlar + Mekinist, Jakavi, Tasigna, Gilenya and Kisqali, as well as Biopharmaceuticals and Emerging Growth Markets.
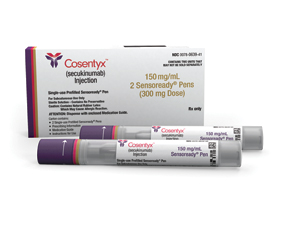
Cosentyx sales in the first six months of 2017 grew to $900 million (+109 percent cc) as the medicine continued its positive launch trajectory with strong growth in psoriatic arthritis (PsA), ankylosing spondylitis (AS) and PsO.

As of the end of May 2017, new weekly prescriptions and the number of US cardiologists that had prescribed Entresto had doubled since 2016 and global access is improving with the heart failure medicine approved in 80 countries.
Entresto sales during the 2017 first half continued to grow ($194 million, +300 percent cc), benefiting from the impact of improved access, U.S. sales-force expansion and reimbursement in Europe.
Promacta/Revolade generated first-half 2017 sales of $385 million, +35 percent cc), driven by continued global uptake as well as growth of the thrombopoietin class for chronic immune thrombocytopenic purpura.
For Tafinlar + Mekinist, January-June 2017 sales rose to $403 million (+28 percent cc), as the performance was driven by double-digit growth across all regions.
With sales of $348 million (+32 percent cc), Jakavi generated continued double-digit growth across all major markets, fueled by myelofibrosis and the launch of the second-line polycythemia vera indication.
Tasigna sales in first-half 2017 improved to $874 million, (+8 percent cc), with solid Q2 growth driven by the U.S. and Emerging Growth Markets.
Novartis says Gilenya exhibited continued growth, as sales rose to about $1.56 billion, accounting for growth of 5 percent at constant currencies compared to first-half 2016.
Sales for Kisqali reached $15 million in the first six months of 2017, as the CDK4/6 inhibitor continues to gain access in the U.S. market. According to Novartis, second-quarter sales were modest with multiple patient access programs available to initiate treatment and bridge to insurance coverage.
First-half 2017 worldwide sales of Biopharmaceuticals rose 17 percent (cc) to $534 million, driven by Zarxio (filgrastim) and Glatopa 20mg (glatiramer acetate) in the United States and double-digit growth in Europe.
Emerging Growth Markets sales advanced 8 percent (cc) year over year to $4.1 billion.
Product Approvals/Launches & Pipeline Updates During 2017
Novartis received the first-ever FDA approval for a CAR-T cell therapy on Aug. 30. Formerly known as CTL019, Kymriah (tisagenlecleucel) suspension for intravenous infusion won marketing clearance for treating patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) that is refractory or in second or later relapse. A novel immunocellular therapy and a one-time treatment, Kymriah uses a patient’s own T cells to fight cancer. This is the first therapy based on gene transfer to gain U.S. regulatory approval.
The first-in-class therapy demonstrated an 83 percent (52/63) overall remission rate in this patient population with limited treatment options and historically poor outcomes. According to Novartis, this novel approach to cancer treatment is the result of a pioneering CAR-T cell therapy collaboration with University of Pennsylvania. During 2012, Novartis and the University of Pennsylvania entered into a worldwide collaboration to further research, develop and commercialize CAR-T cell therapies for the investigational treatment of cancers. Novartis plans additional U.S. and EU submissions for Kymriah before year-end 2017, including applications with the FDA and European Medicines Agency for treating adult patients with r/r diffuse large B-cell lymphoma (DLBCL). Other filings beyond the U.S. and EU markets are anticipated during 2018.
The selective cyclin-dependent kinase inhibitor Kisqali (ribociclib) received U.S. and EU regulatory clearance during 2017. In March, the FDA approved Kisqali, in combination with any aromatase inhibitor, as a treatment for metastatic breast cancer. Novartis says ribociclib in combination with letrozole was added to the National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology (NCCN Guidelines) as a category 1 option for hormone receptor positive, human epidermal growth factor receptor-2 negative (HR+/HER2-) postmenopausal metastatic breast cancer patients.
The European Commission during August approved Kisqali in combination with an aromatase inhibitor for treating postmenopausal women with HR+/HER2- locally advanced or metastatic breast cancer as initial endocrine-based therapy. Kisqali represents the first CDK4/6 inhibitor approved in Europe based on a first-line Phase III study that met its primary endpoint of progression-free survival (PFS) at interim analysis.
Kisqali belongs to a class of medicines that help slow the progression of cancer by inhibiting two proteins called cyclin-dependent kinase 4 and 6 (CDK4/6). These proteins, when over-activated, can allow cancer cells to grow and divide too quickly. Targeting CDK4/6 with enhanced precision may play a role in ensuring that cancer cells do not continue to replicate at an uncontrollable rate.
Kisqali was developed by the Novartis Institutes for BioMedical Research through a research collaboration with Astex Pharmaceuticals.
The FDA’s June approval of Tafinlar (dabrafenib) in combination with Mekinist (trametinib) provides the first targeted treatment in the United States specifically for BRAF V600E mutation-positive metastatic NSCLC. More than 60 percent of treatment-naïve and previously treated patients with BRAF V600E mutant metastatic NSCLC responded to Tafinlar + Mekinist in a pivotal trial. BRAF V600E is an aggressive mutation that may be associated with a poorer prognosis in NSCLC patients. The U.S. regulatory agency had granted Tafinlar + Mekinist Breakthrough Therapy designation during July 2015 for treating patients with advanced or metastatic BRAF V600E mutation-positive NSCLC who received previous treatment with chemotherapy.
Tafinlar + Mekinist was approved by the European Commission during March 2017 for treating patients with BRAF V600 advanced mutation-positive NSCLC.
Combination use of Tafinlar + Mekinist in patients with unresectable or metastatic melanoma who have a BRAF V600 mutation has been approved in the United States, EU, Australia, Canada and other countries.
Tafinlar and Mekinist are additionally indicated in more than 60 countries, including the United States and EU, as single agents for treating patients with unresectable or metastatic melanoma with a BRAF V600 mutation.
Tafinlar and Mekinist target different kinases within the serine/threonine kinase family – BRAF and MEK1/2, respectively – in the RAS/RAF/MEK/ERK pathway, which is implicated in cancers such as NSCLC and melanoma. When Tafinlar is used with Mekinist, the combination has been demonstrated to slow tumor growth more than either drug alone. The Tafinlar + Mekinist combination is being studied in a clinical trial program across a range of tumor types conducted in study centers globally.
The European Commission during September cleared for marketing Rydapt (midostaurin) for two indications in rare, difficult-to-treat cancers. The medicine is approved for use in combination with standard daunorubicin and cytarabine induction and high-dose cytarabine consolidation chemotherapy, and for patients in complete response followed by Rydapt single-agent maintenance therapy, for adults with newly diagnosed acute myeloid leukemia (AML) who are FLT3 mutation-positive. Rydapt was additionally approved for use as monotherapy for treating adults with aggressive systemic mastocytosis (ASM), systemic mastocytosis with associated hematological neoplasm (SM-AHN) or mast cell leukemia.
Rydapt is the first targeted therapy for FMS-like tyrosine kinase 3 (FLT3)-mutated AML and only treatment for three subtypes of SM – collectively known as advanced SM – in the EU, all of which have limited life expectancy and few treatment options. The product represents the first major advancement for treating patients with newly diagnosed FLT3-mutated AML in more than 25 years.
Rydapt is additionally marketed in the United States, Switzerland and Canada. FDA approval was received during April for two indications. The first indication is for treating AML in newly diagnosed patients who are FMS-like tyrosine kinase 3 mutation-positive (FLT3+), as detected by an FDA-approved test, in combination with chemotherapy. Rydapt was additionally cleared to treat adults with advanced SM, which includes ASM, SM-AHN and mast cell leukemia.
Rydapt is an oral, targeted therapy, a type of treatment that interferes with certain pathways involved in the growth, progression, and spread of cancer. The drug inhibits multiple kinases, including FLT3, which help regulate many essential cell processes, interrupting cancer cells’ ability to grow and multiply. The product induces cell death in leukemic cells expressing FLT3 ITD or TKD mutant receptors, or in cells overexpressing FLT3 wildtype receptors.
Rydapt additionally inhibits the activity of the kinase KIT (wild type and D816V mutant), inhibiting mast cell proliferation, survival and histamine release. The medicine also inhibits several other receptor tyrosine kinases such as PDGFR alpha/ß, VEGFR2, and members of the serine/threonine kinase PKC family, inhibiting signaling of the respective growth factors in cells, resulting in growth arrest.
In July, Novartis announced that the European Commission’s Committee for Medicinal Products for Human Use (CHMP) approved a label update for Cosentyx. The label update includes 52-week data from the CLEAR trial showing the long-term superiority of Cosentyx (secukinumab) versus the blockbuster medicine Stelara (ustekinumab) in psoriasis. The updated label additionally includes use of Cosentyx to treat moderate-to-severe scalp psoriasis – one of the most difficult-to-treat forms of psoriasis, which affects 60 million people around the globe. The updated labeling is based on the proven efficacy and consistent safety profile of Cosentyx.
Cosentyx is the only anti-IL17a approved in psoriasis (PsO), psoriatic arthritis (PsA) and ankylosing spondylitis (AS), with strong, sustained efficacy in a majority of patients even after 4 years in PsO, 3 years in PsA, and 2 years in AS.
First-of-its-kind Phase III data were revealed in September demonstrating that Cosentyx delivered high and long-lasting skin clearance in patients with moderate-to-severe plaque psoriasis at 5 years. By specifically targeting interleukin-17A (IL-17A), the medicine addresses the key cytokine involved in the development of psoriasis. IL-17A plays an important role in the pathogenesis of plaque psoriasis, PsA and AS. Inhibiting IL-17A is significant as up to 30 percent of patients with psoriasis may have PsA.
Launched during 2015, Cosentyx is the first fully human IL-17A inhibitor approved for the treatment of psoriasis, PsA and AS. The targeted treatment specifically inhibits the IL-17A cytokine, which plays a significant role in the pathogenesis of plaque psoriasis, PsA and AS.
Cosentyx delivers long-lasting skin clearance, with proven sustainability, safety out to 5 years and convenient once-monthly dosing through a patient-friendly auto injector. The product is also marketed for the most difficult-to-treat types of plaque psoriasis – palmoplantar psoriasis (psoriasis of the hands and feet), scalp psoriasis, and nail psoriasis.
Cosentyx is cleared for marketing in 79 countries for the treatment of moderate-to-severe plaque psoriasis, which includes the EU countries, Japan, Switzerland, Australia, the U.S. and Canada. In Europe, Cosentyx is approved for the first-line systemic treatment of moderate-to-severe plaque psoriasis in adults. In the United States, Cosentyx is available as a treatment for moderate-to-severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy (light therapy).
Additionally, Cosentyx is the first IL-17A inhibitor approved in more than 70 countries for treating active AS and PsA, which includes the EU countries and the United States. Cosentyx is also marketed in Japan for treating PsA and pustular psoriasis.
The European Commission during June approved the expanded use of Zykadia (ceritinib) to include the first-line treatment of patients with advanced non-small cell lung cancer (NSCLC) whose tumors are anaplastic lymphoma kinase (ALK)-positive. The marketing clearance followed a positive opinion granted during May by the CHMP and is applicable to all 28 EU member states as well as Iceland, Lichtenstein, and Norway.
In May, U.S. regulators approved the expanded use of Zykadia to include the first-line treatment of patients with metastatic NSCLC whose tumors are ALK-positive, as detected by an FDA-approved test. Zykadia is an oral, selective inhibitor of ALK, a gene that can fuse with others to form an abnormal “fusion protein” that promotes the development and growth of certain tumors in cancers such as NSCLC.
The European Commission during January approved Votubia (everolimus) dispersible tablets as an adjunctive treatment for patients aged 2 years and older whose refractory partial-onset seizures, with or without secondary generalization, are associated with tuberous sclerosis complex (TSC). Votubia is the first approved pharmacologic therapy in all 28 EU member states, plus Iceland and Norway, specifically for treating refractory partial-onset seizures associated with TSC.
Everolimus is available from Novartis under the trade names Afinitor, Certican and Zortress for use in oncology and transplant patient populations. The drug is exclusively licensed to Abbott and sublicensed to Boston Scientific for use in drug-eluting stents.
During June, the European Medicines Agency accepted the Marketing Authorization Application (MAA) filed by Novartis for AMG 334 (erenumab) for the prevention of migraine. Developed to prevent migraine, erenumab is a fully human monoclonal antibody specifically designed to target and block the Calcitonin Gene-Related Peptide (CGRP) receptor, which is believed to play a critical role in mediating the incapacitating pain of migraine. The drug compound is the only one that is fully human and binds selectively to the CGRP receptor.
Erenumab has been studied in several large worldwide, randomized, double-blind, placebo-controlled trials to evaluate the drug candidate’s safety and efficacy in migraine prevention.
A landmark Phase III study demonstrated that fingolimod significantly reduces relapses in children and adolescents with multiple sclerosis, as revealed by Novartis in September. The PARADIGMS trial is evaluating the safety and efficacy of oral once-daily fingolimod in children and adolescents (ages 10 to 17) with MS. Data demonstrate that oral fingolimod resulted in a significant and clinically meaningful reduction in the amount of relapses (annualized relapse rate) in this patient population over a period of up to two years, compared to interferon beta-1a intramuscular injections. The safety profile of fingolimod was consistent with that evident in other clinical studies, with overall more adverse events reported in the interferon group. PARADIGMS is the first-ever randomized, controlled Phase III trial of a disease-modifying therapy (DMT) in pediatric MS.
Fingolimod is the active chemical in Gilenya, an oral DMT that is highly efficacious at controlling disease activity in relapsing multiple sclerosis (RMS). Gilenya has a reversible lymphocyte redistribution effect targeting focal and diffuse CNS damage caused by MS.
The medicine impacts four key measures of RMS disease activity: relapses, MRI lesions, brain shrinkage (brain volume loss) and disability progression. Gilenya’s effectiveness on all of these measures has been consistently shown in multiple controlled clinical trials and in the real-world setting. Studies have demonstrated the drug’s safety and high efficacy to be sustained over the long term, showing that switching to Gilenya treatment as early in the disease course as possible can be beneficial in helping to preserve individuals’ function.
Gilenya is FDA-approved for the first-line treatment of relapsing forms of MS in adults and in the EU for adult patients with highly-active relapsing-remitting MS (RRMS) defined as either high disease activity despite treatment with at least one DMT, or rapidly evolving severe RRMS.
Novartis during August announced analysis published in The Lancet showing ACZ885 reduced lung cancer mortality by 77 percent in the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). A review of blinded, pre-planned oncology safety analyses revealed a 77 percent reduction in lung cancer mortality and a 67 percent reduction in lung cancer cases in patients treated with 300 milligrams of ACZ885.
The Phase III trial studied the role of the interleukin-1ß antibody ACZ885 in people with a prior heart attack and inflammatory atherosclerosis as measured by high-sensitivity C-reactive protein (hsCRP), a known marker of inflammation, at levels of >=2mg/L. CANTOS is the first Phase III study to support a long-established hypothesis from pre-clinical models that inhibition of IL-1ß impacts cancer incidence and mortality. The CANTOS cardiovascular trial met its primary endpoint as published in The New England Journal of Medicine, validating that the anti-inflammatory agent impacts cardiovascular risk reduction.
Novartis intended to discuss lung cancer hypothesis with regulatory authorities and begin evaluation in other Phase III confirmatory trials.
Novartis in June reported top-line results from CANTOS investigating the efficacy, safety and tolerability of ACZ885 in combination with standard of care in people with a prior heart attack and inflammatory atherosclerosis. The CANTOS trial met the primary endpoint, demonstrating that when used in combination with standard of care, ACZ885 reduces the risk of major adverse cardiovascular events (MACE) – a composite of cardiovascular death, non-fatal myocardial infarction and non-fatal stroke – in patients with a previous heart attack and inflammatory atherosclerosis.
With more than 10,000 patients enrolled in the clinical trial during the last six years, CANTOS is one of the largest and longest-running studies in Novartis’ history.
A selective, high-affinity, fully human monoclonal antibody, ACZ885/canakinumab inhibits IL-1ß – a key cytokine in the inflammatory pathway known to drive the continued progression of inflammatory atherosclerosis. ACZ885 works by blocking the action of IL-1ß for a sustained period of time, therefore inhibiting inflammation resulting from its over-production. ACZ885 is the first investigational treatment that has shown that selectively targeting inflammation significantly reduces cardiovascular risk.
Biosimilar Progress During 2017
As a worldwide leader in biosimilars, Sandoz had five biosimilars approved in Europe as of September, including versions of some of the world’s leading blockbuster biologics.
The European Commission during June approved Rixathon (biosimilar rituximab) for use in all indications of the reference medicine, Roche’s blockbuster medicine MabThera, for treating blood cancers and immunological diseases. Rixathon was cleared for treating non-Hodgkin’s lymphoma (follicular lymphoma and diffuse large B-cell lymphoma) and chronic lymphocytic leukemia, as well as immunological diseases including rheumatoid arthritis, granulomatosis with polyangiitis, and microscopic polyangiitis.
The FDA accepted the Biologics License Application under the 351 (k) pathway for a proposed biosimilar to the reference medicine Rituxan (rituximab), as reported by Novartis in September. Rituxan is the U.S. branded version of MabThera.
Another June EU biosimilar approval for Sandoz came in the form of Erelzi (biosimilar etanercept). Erelzi was approved for use in all indications of the reference medicine Enbrel, which is marketed for treating immunological diseases and is one of the top-selling prescription medicines worldwide.
Erelzi was approved for rheumatoid arthritis, axial spondyloarthritis (ankylosing spondylitis and non-radiographic axial spondyloarthritis), plaque psoriasis, and psoriatic arthritis, juvenile idiopathic arthritis and pediatric plaque psoriasis. Erelzi is available in a pre-filled syringe and an auto-injector pen called SensoReady, which has been designed for patient safety, comfort, and convenience.
Sandoz revealed new data in September on its proposed biosimilar adalimumab, which is marketed as Humira, the world’s best-selling prescription medicine. Data from a long-term study of patients continuously treated with the proposed biosimilar or the reference medicine demonstrate that efficacy and safety profiles of the two medicines match throughout 51 weeks of treatment in patients with moderate-to-severe chronic plaque psoriasis.
Sandoz’s proposed biosimilar adalimumab is undergoing review by the European Medicines Agency for treating several immunological diseases. Novartis announced on May 30 that the EMA accepted the Marketing Authorization Application for biosimilar Humira.
On that same date, the company additionally reported that the EMA accepted for regulatory review the Marketing Authorization Application for a biosimilar version of Janssen’s Remicade (infliximab), which is used to treat immunological diseases. Sandoz is seeking marketing clearance for biosimilar adalimumab and infliximab for use in all indications of their respective reference medicines.
